Xơ nang
| Bệnh xơ nang | |
|---|---|
| Tên khác | Mucoviscidosis / Bệnh nhày nhớt trẻ sơ sinh |
 | |
| Khoa/Ngành | Di truyền y học, Khoa hô hấp |
| Triệu chứng | Khó thở, nhiều đờm, v.v. |
| Khởi phát | Triệu chứng nhận biết được khi bệnh nhân khoảng 6 tháng tuổi[1] |
| Diễn biến | Dai dẳng, kéo dài |
| Nguyên nhân | Di truyền Mendel do alen lặn |
| Phương pháp chẩn đoán | Xét nghiệm di truyền |
| Điều trị | Kháng sinh, Enzyme tụy (dược phẩm), Cấy ghép phổi |
| Tiên lượng | Tuổi thọ tối đa 42 đến 50 tuổi (ở các nước phát triển) |
| Dịch tễ | 1 / 3,000 ở Bắc Âu |
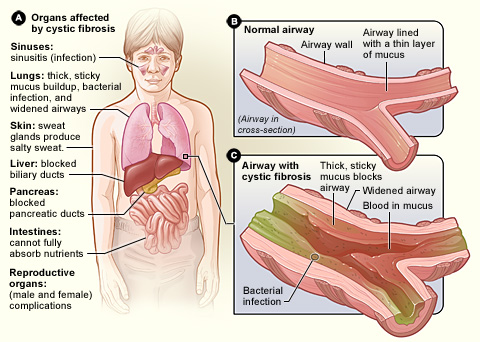
Xơ nang là một bệnh do rối loạn di truyền gây ra biến đổi bất thường của các tuyến ngoại tiết, chủ yếu là các tuyến tụy, gan và thận ; từ đó có thể tác động xấu đến phổi và ruột. Nói chung, bệnh diễn biến không nhanh, nhưng thường có thể dẫn đến bệnh phổi mãn tính, suy gan mật, tăng điện giải tuyến mồ hôi, tỉ lệ tử vong ở trẻ sơ sinh khá cao.[2][3][4]
Tên bệnh này tương ứng với thuật ngữ tiếng Anh là cystic fibrosis (viết tắt là CF), là bệnh di truyền do cặp alen lặn đột biến gây ra, nên trẻ sinh ra khoảng 6 tháng mang cặp alen lặn này đã biểu hiện bệnh. Trong Nhi khoa, tên bệnh CF này còn được gọi là bệnh nhày nhớt.[5]
Tổng quan
[sửa | sửa mã nguồn]- CF (xơ nang hay cystic fibrosis) ban đầu biểu hiện ở khó thở và ho ra nhiều chất nhầy do nhiễm trùng phổi thường xuyên. Lâu dài có thể thêm các triệu chứng như nhiễm trùng xoang, ngón tay và ngón chân dùi trống, và vô sinh ở hầu hết nam giới. Những người khác nhau có thể có mức độ khác nhau của các triệu chứng trên.
- CF khá phổ biến nhất trong số những người có nguồn gốc Bắc Âu và tần số biểu hiện bệnh ở các cộng đồng người này là khoảng 1/3000 trẻ sơ sinh, nghĩa là khoảng 1/25 người trong quần thể là thể mang (thể dị hợp). Ở cộng đồng người châu Phi và châu Á thì tần số thấp hơn nhiều.
- Bệnh CF này lần đầu tiên được công nhận là một bệnh thực thể nhờ nghiên cứu của Dorothy Andersen vào năm 1938, với các mô tả phù hợp với thể bệnh được mô tả gần giống như thế ít nhất là từ năm 1595.[6] Cái tên "xơ nang" dùng để chỉ các loại xơ và nang hình thành trong tuyến tụy, đặc trưng cho bệnh này.[6][7]
- Hiện nay không có phương pháp điều trị triệt để được bệnh CF này.[8] Các biện pháp điều trị chủ yếu là điều trị triệu chứng, như chống nhiễm trùng phổi bằng kháng sinh, thường là azithromycin được sử dụng lâu dài. Khí dung muối hypertonic và salbutamol cũng có thể tác dụng điều trị ở một số người bệnh. Ghép phổi có thể là một lựa chọn nếu chức năng phổi suy giảm nghiêm trọng.[9] Thay thế enzyme tụy và bổ sung vitamin tan trong chất béo rất quan trọng, đặc biệt là ở người trẻ.[10] Tuổi thọ trung bình của người bệnh là từ 42 đến 50 năm ở các nước phát triển.[11][12] Biến đổi nghiêm trọng ở phổi là nguyên nhân gây tử vong ở 80% người bệnh.
Nguyên nhân
[sửa | sửa mã nguồn]
- Gen bình thường (không gây bệnh CF) là alen trội, định vị trên cánh dài của nhiễm sắc thể số 7. Alen này mã hóa một prôtêin liên kết màng gọi là CFTR (cystic fibrosis transmembrane conductance regulator, tức là "điều hòa độ dẫn xuyên màng xơ nang"). Gen CFTR cung cấp các thông tin để tổng hợp nên một sản phẩm gọi là "prôtêin điều hòa độ dẫn truyền qua màng của bệnh xơ nang". Prôtêin CFTR có chức năng là kênh vận chuyển xuyên màng tế bào, sản xuất các chất tiết như chất nhờn, mồ hôi, nước bọt, nước mắt và cả các enzym ngoại tiết. Kênh vận chuyển các hạt mang điện tích âm được gọi là ion clorua vào và ra khỏi tế bào. Sự vận chuyển của các ion clorua giúp kiểm soát sự di chuyển của nước trong các mô, điều này cần thiết cho việc sản xuất chất nhầy loãng và chảy tự do. Chất nhầy là một chất trơn có tác dụng bôi trơn và bảo vệ niêm mạc của đường hô hấp, hệ tiêu hóa, hệ sinh sản cũng như nhiều mô và cơ quan mô khác.[13]
- Khi gen CFTR bị đột biến, thì đột biến gen phổ biến nhất là đột biến F508del (chiếm khoảng 86% các alen đột biến) ở trạng thái lặn so với gen bình thường. Đột biến lặn của CFTR làm cho kênh clorua điều hòa cAMP, điều hòa vận chuyển clorua và natri qua màng biểu mô bị rối loạn. Vì đột biến này là lặn (ở đây kí hiệu là a), nên bệnh chỉ biểu hiện ở thể đồng hợp (aa). Thể đồng hợp aa này thường là con của cặp vợ chồng bình thường, nhưng đều là thể mang (tức bố và mẹ = Aa Aa). Cặp alen lặn đồng hợp dẫn đến việc tạo ra chất nhờn đặc hơn và dính hơn bình thường. Chất nhầy này khó khạc ra ngoài phổi. Điều này có thể gây khó thở và dẫn đến nhiễm trùng phổi nặng. Chất nhầy cũng can thiệp vào chức năng tuyến tụy bằng cách ngăn chặn các enzym phân hủy thức ăn đúng cách. Hệ quả là các vấn đề về tiêu hóa, có khả năng dẫn đến suy dinh dưỡng. Sự đặc quánh của chất nhầy này cũng có thể gây vô sinh nam do làm tắc ống dẫn tinh, hoặc ống dẫn tinh trùng từ tinh hoàn đến niệu đạo. CF nghiêm trọng, với những hậu quả có thể đe dọa đến tính mạng. Nguyên nhân tử vong phổ biến nhất ở những người bị CF là suy hô hấp.[3][4][14]
- Thể dị hợp tử (Aa) không biểu hiện bệnh, nhưng có thể có những bất thường tinh vi về vận chuyển điện giải qua tế bào biểu mô nhưng không bị ảnh hưởng về mặt lâm sàng. Người ta đã phân loại các đột biến CFTR thành năm lớp dựa trên sự thay đổi đột biến ảnh hưởng đến chức năng hoặc quá trình sản xuất prôtêin CFTR . Bệnh nhân có đột biến gen I, II hoặc III được xem là có kiểu gen nặng hơn dẫn đến chức năng CFTR rất ít hoặc không có trong khi bệnh nhân có 1 hoặc 2 đột biến ở cấp độ IV hoặc V được xem là có kiểu gen nhẹ hơn dẫn đến chức năng CFTR còn lại . Tuy nhiên, không có mối liên quan chặt chẽ giữa các đột biến đặc hiệu và biểu hiện bệnh cụ thể, vì vậy việc kiểm tra lâm sàng (ví dụ chức năng của cơ quan) thay vì kiểu gen là hướng dẫn tốt hơn để tiên lượng.[2][15][16]
Nguồn trích dẫn
[sửa | sửa mã nguồn]- ^ Allen JL, Panitch HB, Rubenstein RC (2016). Cystic Fibrosis (bằng tiếng Anh). CRC Press. tr. 92. ISBN 9781439801826. Lưu trữ bản gốc ngày 8 tháng 9 năm 2017.
- ^ a b Beryl J. Rosenstein. “Xơ nang”.
- ^ a b “Everything you need to know about cystic fibrosis”.
- ^ a b Massie J, Delatycki MB (tháng 12 năm 2013). “Cystic fibrosis carrier screening”. Paediatric Respiratory Reviews. 14 (4): 270–5. doi:10.1016/j.prrv.2012.12.002. PMID 23466339.
- ^ “Bệnh nhầy nhớt ở trẻ sơ sinh”.
- ^ a b Hodson, Margaret; Geddes, Duncan; Bush, Andrew biên tập (2012). Cystic fibrosis (ấn bản thứ 3). London: Hodder Arnold. tr. 3. ISBN 978-1-4441-1369-3. Lưu trữ bản gốc ngày 8 tháng 9 năm 2017.
- ^ Andersen DH (1938). “Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study”. Am J Dis Child. 56 (2): 344–399. doi:10.1001/archpedi.1938.01980140114013.
- ^ Lỗi chú thích: Thẻ
<ref>sai; không có nội dung trong thẻ ref có tênMas2013 - ^ Lỗi chú thích: Thẻ
<ref>sai; không có nội dung trong thẻ ref có tênO2009 - ^ Warnock, L; Gates, A (ngày 21 tháng 12 năm 2015). “Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis”. The Cochrane Database of Systematic Reviews (12): CD001401. doi:10.1002/14651858.CD001401.pub3. PMID 26688006.
- ^ Ong, T; Ramsey, BW (ngày 15 tháng 9 năm 2015). “Update in Cystic Fibrosis 2014”. American Journal of Respiratory and Critical Care Medicine. 192 (6): 669–75. doi:10.1164/rccm.201504-0656UP. PMID 26371812.
- ^ Nazareth, D; Walshaw, M (tháng 10 năm 2013). “Coming of age in cystic fibrosis - transition from paediatric to adult care”. Clinical Medicine. 13 (5): 482–6. doi:10.7861/clinmedicine.13-5-482. PMC 4953800. PMID 24115706.
- ^ “CFTR gene”.
- ^ Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D (tháng 1 năm 2004). “Cystic fibrosis adult care: consensus conference report”. Chest. 125 (1 Suppl): 1S–39S. CiteSeerX 10.1.1.562.1904. doi:10.1378/chest.125.1_suppl.1S. PMID 14734689.
- ^ Buckingham, Lela (2012). Molecular diagnostics fundamentals, methods, and clinical applications (ấn bản thứ 2). Philadelphia: F.A. Davis Co. tr. 351. ISBN 978-0-8036-2975-2. Lưu trữ bản gốc ngày 8 tháng 9 năm 2017.
- ^ Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D (2004). “Cystic fibrosis adult care consensus conference report”. Chest. 125 (90010): 1–39. CiteSeerX 10.1.1.562.1904. doi:10.1378/chest.125.1_suppl.1S. PMID 14734689.