Adding bond types & orders to standard amino acid bonds and cys-cys bonds #3770
Conversation
|
Thanks! That looks really interesting. How did you generate the updated One concern I have is that setting bond types from PDB files will always be inconsistent. Standard bonds will be marked correctly, but any nonstandard ones from CONECT records (anything other than proteins, nucleic acids, and water, as well as covalent modifications to proteins) won't be. I worry that people will see bond types being present, assume that means all bonds have specified types, and write code like this. for bond in topology.bonds():
if bond.type == 'Single':
# Do something that's supposed to happen for every single bond, but actually won't.Does anyone have thoughts on this? |
|
@peastman my best answer would be to have them have |
|
A few points from the OpenFF side:
|
…/github.com/RiesBen/openmm into Adding_bondTypes_Orders_to_stdBonds_CCBonds
|
Hi all, @peastman I generated the .xml file semi-automatically based on Lehninger Principles of Biochemistry (text-book) @j-wags @richardjgowers I like the idea of having None bond-order for undefined and maybe "Undefined" for bond-type? @j-wags if formal charges should be added, I think much more of the code needs to be significantly modified.I would like to suggest to split this into a second PR? |
What about other residues that have multiple protonation states? Do they need special treatment too?
That's how it works right now. The difference is that currently all of them are undefined, because we don't read any file formats that provide bond orders. There are two possible problems we need to consider:
Both of these are concerns, but of course the second one is a much bigger concern. As you observed, simply adding or removing a hydrogen can change nearby bonds. Same with any covalent modification. How confident are we that any bond orders that get defined will be correct? For what it's worth, tools like RdKit and OpenBabel try to automatically infer bond orders and formal charges, and in my experience neither one of them is reliable. |
|
Just commenting regarding another use case that could potentially be enabled here: If we can get to the point where we have {atom elements and formal charges, and bonds with Kekulized integral bond orders} will be represented in |
|
For reference, here's the code OpenBabel uses to infer bond orders. It uses a lot of geometric criteria to identify special cases. RDKit's PDB reader uses a method more like the one here, with a table of which bonds in standard residues are double bonds. Though the code is a bit horrifying to look at. And of course, that only works for standard residues, nothing else. |
|
I'm going to meet with @RiesBen and @richardjgowers tomorrow to chat more about OpenFF's and OpenFE's needs regarding PDB loading, and whether it would be best for the code to live in OpenMM or elsewhere. But based on a quick discussion today, it seems like @richardjgowers and I are in agreement that formal charge is needed to unambiguously identify a chemical species. I think my confusion was around whether this PR was intended to support complete chemical identification when loading from PDB. My major concern in this discussion was that, if we wanted this PR to enable identifying chemical species from PDB files, we'd need to implement formal charge assignment as well, and implementing atomic formal charge in OpenMM's Topology class would be a significant API/behavior change. So my concerns about this PR were along the lines of "if Peter is as resistant to behavior changes as I am, and we know we'll eventually want formal charge assigners as well, then it should all go in one PR". @RiesBen and @richardjgowers' motivation, if I understand this correctly, is "we've already got all of the bond order assignment stuff lined up in a fork that we're using, we may as well upstream it since it's not an API change". This also makes sense - OpenMM Topologies already have a field for bond order+type information, so there's little harm in filling it in. So, if we're in agreement that the goal of this PR isn't "fill in complete chemical information for molecules loaded from PDB", then my major concern about this functionality is resolved! |
|
That seems fine. My main concern at this point is just about correctness. How confident are we that the bond information it fills in will always be accurate, including for different protonation states, different types of chain termination, and proteins with post-translational modifications? |
|
The correct long-term solution here---which OpenFF has already started to roll out---is to use the Chemical Components Dictionary as the canonical reference of residues described in the PDB, since the formal charges and bond orders there for all acceptable variants in the PDB are provided and validated. |
|
It's correct as long as 1) your file only contains things that have been submitted to the PDB, and 2) it refers to everything with the correct names. In practice, a lot of PDB files don't meet those requirements. |
(1) PDB and mmCIF formats were developed to represent things that came from the PDB. A different format is really necessary for specifying residues or molecules not contained in the PDB---for the exact reason that the PDB does not provide sufficient information to define the chemistry. Their use for representing other things is a misuse and something to provide "best effort" support for, but not something that should preclude supporting their primary intended purpose. (2) The PDB enforces this naming convention, but you could always use the molecular graph to match residues. The difficulty is that you would need to know how to transform the complete molecules in the CCD into common residues of those molecules (which are only parts of molecules)---a problem that the OpenFF folks have likely examined in much more detail. |
Even if you assume you're dealing with a file straight from RCSB, there generally isn't enough information to determine what you want. That's because they usually don't have hydrogens, which makes protonation states ambiguous. That in turn means that bond orders and formal charges are ambiguous. It's impossible to assign them when you load the file. You have to wait until hydrogens are added. |
|
@peastman regarding PTMs, the standard stuff like phosphorylation, sulfatization, glycolization, etc. should not break the current implementation, if we would estimate the bond-orders/add them to the residue.xml, as they should be well behaved. Example(here serine without phosphate would have a -1 fc, with it would be neutral): Alternative problems may be coming from different oxidation states of many-electron metals (Fe, etc.). @peastman do you have a specific case in mind, that concerns you? :) |
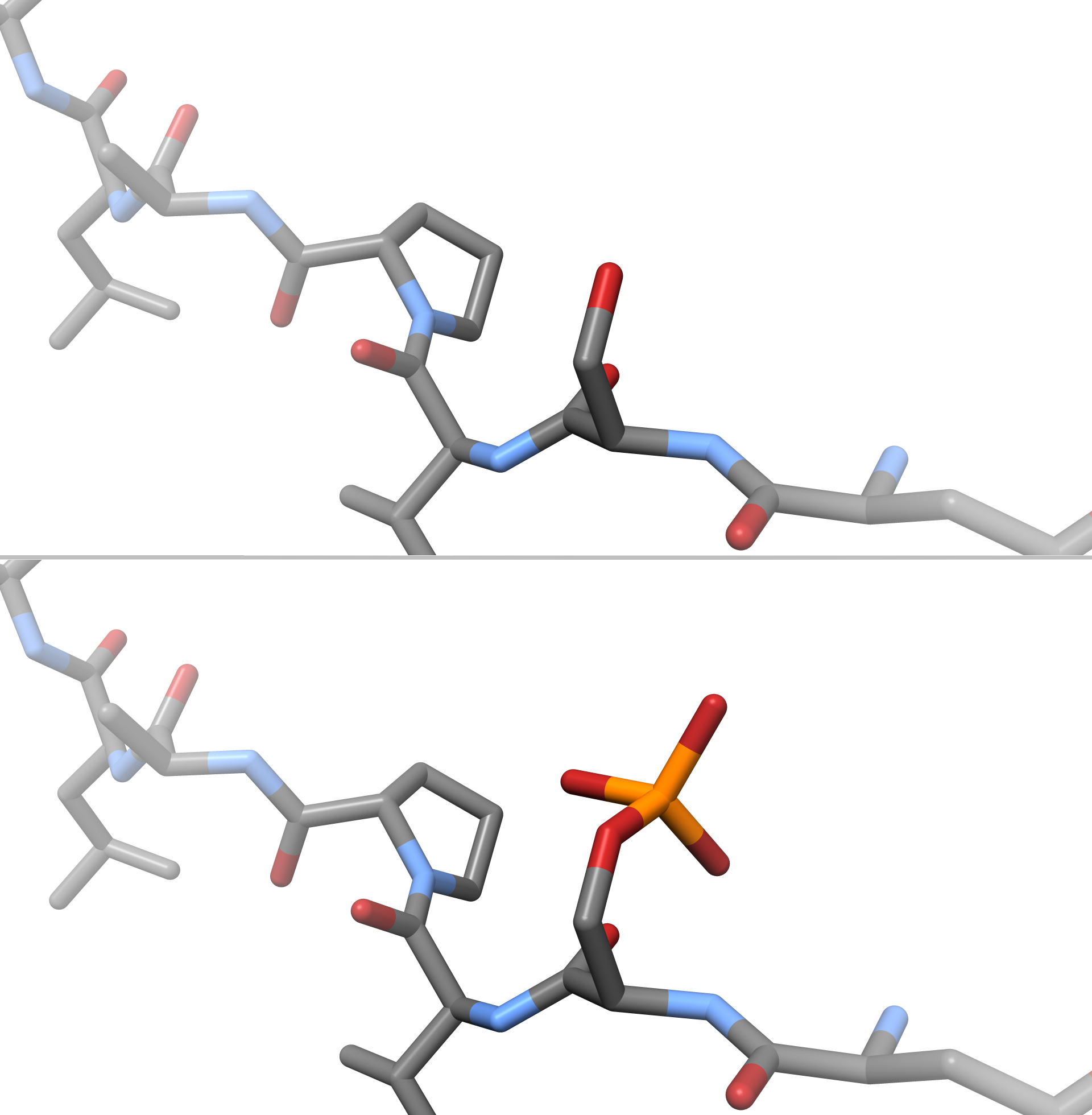
I worry about the cases I know about, but also the ones I don't know about! :) This feature will definitely need a good set of tests. As a starting point, can we verify that the following cases are all handled correctly?
We should check all the variants supported by |
|
Regarding the PTMs this is a cool collection of 'frequent' PTMs. (I guess in a lot of modelling approaches they are still rare) I think besides the ubiquitin, they could be easily realized with templates like the residue.xml already contains (either recognize them separate from amino acids (AA) or as modified AAs - only a couple of AAs are affected), consequently the current implementation would not need to change. hm I'm off now for some time (3 Weeks), but we can discuss the next steps, when I'm back. :) |
|
There's a lot of design and testing still needed first. See the discussion above, for example #3770 (comment). |
|
@peastman I made a pdb with every aminoacid variant, sequence is GLY-ASH-ASP-CYS-CYX-GLH-GLU-HID-HIE-HIP-HIN-LYN-LYS. Also, to check different caps, GLY-GLY and ACE-GLY-GLY-NME. These are made in pymol. Unfortunately these don't load into openmm 7.7 out of the box, even without the changes in this PR. Code: Result: The pdbs seem fine to me by looking through them. Could you help figure out what it is that openmm doesn't like about them? (Possible clue: |
|
@peastman Once we do get variants.pdb loading, how can we check the bond orders and types? We could inspect those manually and set up a regression test. Alternatively, we could compare with the bond orders when the same pdb is read in by oechem. |
|
@aizvorski I think a better gold set would be the PDB Chemical Components Dictionary: https://www.wwpdb.org/data/ccd |
|
Your file uses nonstandard naming. See https://github.com/openmm/openmm/wiki/Frequently-Asked-Questions#template for a discussion of what causes this error, and why naming matters.
Agreed, that seems like a good reference. |
@richardjgowers Good reference, thanks. Do you mean the "Protonation Variants Companion Dictionary" within that? The big Chemical Component Dictionary contains a whole bunch of ligands which is probably overkill, although it also includes common modified residues like PTR and TPO. I had a peek and it includes both aa in the normal protonation state and a wide variety of other states, including many which OpenMM wouldn't handle currently (and also for which I think there may not be Amber forcefield parameters). Fully supporting all of those seems like a tall order, and a much bigger feature project than just adding bond orders. There are bond orders in there, so it actually seems feasible to write a converter from https://files.wwpdb.org/pub/pdb/data/monomers/aa-variants-v1.cif to openmm/app/data/residues.xml |
Hmm, it seems so, even though it comes straight from Pymol (Pymol bug?). I ran it through pdbfixer which added extra hydrogens, ie both H02 and H2. Manually fixing the names in glygly.pdb works though. The error message is wrong/confusing: "The set of atoms matches NGLY, but the bonds are different" - that doesn't say anything about nonstandard atom names, or which atoms have the nonstandard names, or how to fix the problem. Why does the code think the bonds are different, as opposed to atom names? Which bonds? |
At least initially, I think it's sufficient to only support the variants used by
See https://github.com/openmm/openmm/wiki/Frequently-Asked-Questions#template for an explanation of what's going on. |
Hi openmm,
over at openfe/gufe, we wanted to use the PDBFile parser for reading in protein pdbs here! :) But we want to use gufe with openff and therefore need to have the bond-orders. This is currently not possible but easily doable with minor changes to your implementation (as nearly everything is already there). I connected the loose ends and want to suggest these small, non-invasive changes to the topology class in openmm.
Changes:
Please let me know what you think! :)