Enfermedad de Alzheimer
La enfermedad de Alzheimer (EA), denominada demencia senil de tipo alzhéimer (DSTA), o simplemente alzhéimer[4] es una enfermedad neurodegenerativa, producto de un proceso de neurodegeneración y que se manifiesta como deterioro cognitivo y trastornos conductuales. Se caracteriza en su manera típica por una pérdida de la memoria inmediata y de otras capacidades mentales (tales como las capacidades cognitivas superiores), a medida que mueren las células nerviosas (neuronas) y se atrofian diferentes zonas del cerebro. La enfermedad suele tener una duración media aproximada —después del diagnóstico— de 10 años,[5] aunque esto puede variar en proporción directa con la severidad de la enfermedad al momento del diagnóstico.
| Enfermedad de Alzheimer | ||
|---|---|---|
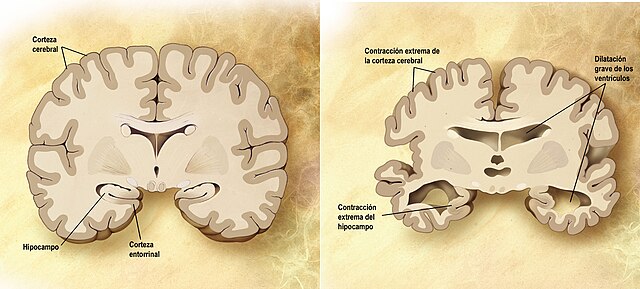 Comparación entre un cerebro normal (izquierda) y un cerebro afectado de alzhéimer (derecha). | ||
| Especialidad | neurología | |
| Síntomas | deterioro cognitivo, pérdida de la memoria, cambios en la personalidad, deterioro de la marcha, desorientación, cambios de humor[1] | |
| Inicio habitual | A partir de los 65 años.[2] | |
| Duración | crónica[1] | |
| Causas | desconocidas, factores genéticos[1] | |
| Diagnóstico | basada en los síntomas y pruebas cognitivas[1] | |
| Tratamiento | sintomático[1] | |
| Medicación | Inhibidores de la acetilcolinesterasa[1] | |
| Pronóstico | Esperanza de vida entre 3-9 años tras diagnóstico[3] | |
| Sinónimos | ||
| ||

La enfermedad de Alzheimer es la forma más común de demencia, es incurable y terminal, y aparece con mayor frecuencia en personas mayores de 65 años de edad,[6] aunque también en raros casos puede desarrollarse a partir de los 40 años de edad.
Muchas personas experimentan olvidos o retrasos leves de memoria, que son parte del proceso normal de envejecimiento. La mayoría de las personas tienen dificultades ocasionales para recordar una palabra o el nombre de alguien. Sin embargo, una persona con la enfermedad de Alzheimer u otros tipos de demencia, encontrará estos síntomas cada vez más frecuentes y graves.
Los signos que indican la enfermedad de Alzheimer pueden incluir:
- Cambios en la personalidad
- Deterioro en la capacidad de movimiento o al caminar
- Dificultad para comunicarse
- Bajo nivel de energía
- Pérdida de memoria
- Cambios de estado de ánimo
- Problemas de atención y orientación
- Incapacidad de resolver operaciones aritméticas sencillas
Los síntomas como una entidad nosológica definida fueron identificados por el psiquiatra alemán Emil Kraepelin,[7] mientras que la neuropatología característica fue observada por primera vez por el psiquiatra y neurólogo alemán Alois Alzheimer en 1906.[8][9][10]
Así pues, el descubrimiento de la enfermedad fue obra de ambos psiquiatras, que trabajaban en el mismo laboratorio. Sin embargo, dada la gran importancia que Kraepelin daba a encontrar la base neuropatológica de los desórdenes psiquiátricos, decidió, en 1910, nombrar a la enfermedad Alzheimer en honor a su compañero.[11]
Por lo general, el síntoma inicial es la inhabilidad de adquirir nuevos recuerdos, pero suele confundirse con actitudes relacionadas con la vejez o el estrés.[12]
Ante la sospecha de alzhéimer, el diagnóstico se realiza con evaluaciones de conductas cognitivas, así como neuroimágenes, si están disponibles.[13]
A medida que progresa la enfermedad, aparecen confusión mental, irritabilidad y agresión, cambios del humor, trastornos del lenguaje, pérdida de la memoria de corto plazo y una predisposición a aislarse a medida que declinan los sentidos del paciente.[12][14]
Gradualmente se pierden las funciones biológicas, que finalmente conllevan a la muerte.[15]
El pronóstico para cada individuo es difícil de determinar. El promedio general es de siete años;[16] menos del 3 % de los pacientes viven más de 14 años después del diagnóstico.[17]
La causa de la enfermedad de Alzheimer permanece desconocida, aunque las últimas investigaciones parecen indicar que están implicados procesos de tipo priónico.[18] Las investigaciones suelen asociar la enfermedad a la aparición de placas seniles y ovillos neurofibrilares.[19] Los tratamientos actuales ofrecen moderados beneficios sintomáticos, pero no hay tratamiento que retrase o detenga el progreso de la enfermedad.[20] No obstante, casos preliminares de asociación de demencia por alzhéimer con la enfermedad celíaca mostraron la mejoría con el seguimiento de una dieta sin gluten.[21] En la actualidad, el diagnóstico exacto solo se consigue post mortem, por lo que existe un gran interés en encontrar marcadores genéticos que permitan una detección temprana de esta enfermedad que sería más fácil de tratar que en los estadios más avanzados.
Para la prevención del alzhéimer, se han sugerido varios hábitos conductuales, pero no hay evidencias publicadas que destaquen los beneficios de esas recomendaciones, incluyendo la estimulación mental y la dieta equilibrada.[22]
El papel que juega el cuidador del sujeto con alzhéimer es fundamental,[23] aun cuando las presiones y la demanda física de esos cuidados pueden llegar a ser una gran carga personal.[24][25][26]
El Día Internacional del Alzheimer se conmemora el 21 de septiembre, fecha elegida por la OMS y la Federación Internacional de Alzhéimer, en la cual se llevan a cabo actividades en diversos países para concienciar y ayudar a prevenir la enfermedad.
La Organización Mundial de la Salud (OMS) realizó en 2015 su Primera Conferencia Ministerial de la OMS sobre la Acción Mundial contra la Demencia.
Historia
editarMédicos griegos y romanos asociaron la vejez con la demencia.[10] Pero no fue hasta 1901 cuando el psiquiatra alemán Alois Alzheimer identificó el primer caso de lo que se conoce hoy como enfermedad de Alzheimer en una mujer de 51 años de edad; esta mujer se llamaba Auguste Deter. El investigador hizo seguimiento de su paciente hasta su muerte en 1906, y entonces pudo observar el cerebro. Después de este momento, pudo reportar públicamente por primera vez el caso.[27]
Uno de los primeros síntomas de una mujer de 51 años fue un fuerte sentimiento de celos hacia su marido. Pronto mostró progresivos fallos de memoria, no podía encontrar el camino a casa, arrastraba objetos sin sentido, se escondía o a veces pensaba que otras personas querían matarla, de forma que empezaba a gritar.Durante su internamiento, sus gestos mostraban una completa impotencia. Estaba desorientada en tiempo y espacio. De cuando en cuando decía que no entendía nada, que se sentía confusa y totalmente perdida. A veces consideraba la llegada del médico como la visita de un funcionario y pedía perdón por no haber acabado su trabajo, mientras que otras veces comenzaba a gritar por temor a que el médico quisiera operarla. En ocasiones lo despedía completamente indignada, chillando frases que indicaban su temor a que el médico quisiera herir su honor. De vez en cuando estaba completamente delirante, arrastrando las mantas de un lado a otro, llamando a su marido y a su hija, y con aspecto de tener alucinaciones auditivas. Con frecuencia gritaba durante horas y con una voz horrible.
La regresión mental avanzó gradualmente. Tras cuatro años y medio de enfermedad, la paciente falleció. Al final estaba completamente apática y confinada a la cama, donde adoptaba una posición fetal.
Tras la muerte de la mujer, Alzheimer le examinó el cerebro con un microscopio. Anotó las alteraciones de las «neurofibrillas», elementos del citoesqueleto teñidos con una solución de plata.
La preparación de plata de Bielschowsky mostró cambios muy característicos de las neurofibrillas. Sin embargo, en el interior de la célula de aspecto normal se podía observar una o varias fibras únicas que eran prominentes por su grosor y su impregnabilidad. En una etapa más avanzada, muchas fibrillas dispuestas en paralelo mostraban los mismos cambios. Luego se acumulaban formando densos haces y gradualmente avanzaban hacia la superficie de la célula. Algunas veces, el núcleo y el citoplasma desaparecían, y solo un conjunto de haces de fibrillas indicaba el lugar donde había existido una neurona. Como estas fibras podían ser teñidas con tinciones diferentes de las neurofibrillas normales, tenía que haberse producido una transformación química de la sustancia fibrilar. Esta podría ser la razón por la que las fibrillas sobrevivían a la destrucción de la célula. Parece que la transformación de las fibrillas coincide con el almacenamiento de un producto patológico todavía no bien conocido del metabolismo de la neurona. Alrededor de un cuarto o un tercio de todas las neuronas de la corteza cerebral mostraban esas alteraciones. Numerosas neuronas, especialmente en las capas celulares altas, habían desaparecido totalmente.
Durante los siguientes cinco años, la literatura médica reportó al menos once casos similares, algunos de ellos utilizando ya el término «enfermedad de Alzheimer».[10] La enfermedad fue categorizada por primera vez por Emil Kraepelin después de la supresión de algunos elementos clínicos concomitantes como delirios y alucinaciones, así como características histológicas irrelevantes para la enfermedad, como los cambios arterioscleróticos, los cuales figuraban en el informe original sobre Auguste D.[30]
En la octava edición de su libro de texto de psiquiatría, publicado en 1910, incluyó a la enfermedad de Alzheimer, denominada también por Emil Kraepelin «demencia presenil», como un subtipo de demencia senil.[31]
Durante la mayor parte del siglo XX, el diagnóstico del Alzhéimer se reservaba para las personas entre las edades de 45 y 65 años con síntomas de demencia. La terminología ha cambiado desde 1977, cuando, en una conferencia sobre Alzhéimer, se llegó a la conclusión de que las manifestaciones clínicas y patológicas de la demencia presenil y senil eran casi idénticas, aunque los autores también agregaron que ello no descartaba la posibilidad de que tuviesen causas diferentes.[32]
Esto, a la larga, conllevó a que se hiciera el diagnóstico del alzhéimer independientemente de la edad.[33]
El término demencia senil del tipo alzhéimer fue empleado durante un tiempo para describir el trastorno en aquellos mayores de 65 años de edad, mientras que la enfermedad clásica de Alzheimer se reservaba para los de edades menores. Finalmente, el término enfermedad de Alzheimer se aprobó oficialmente en la nomenclatura médica para describir a individuos de todas las edades con un patrón de síntomas: característica, curso de la enfermedad y neuropatología comunes.[34]
Epidemiología
editar| Edad | Incidencia (nuevos casos) por cada mil personas |
|---|---|
| 65-69 | 3 |
| 70-74 | 6 |
| 75-79 | 9 |
| 80-84 | 23 |
| 85-89 | 40 |
| 90- | 69 |
La incidencia en estudios de cohortes muestra tasas entre 10 y 15 nuevos casos cada mil personas al año para la aparición de cualquier forma de demencia y entre 5 a 8 para la aparición del alzhéimer.[35][36]
Es decir, la mitad de todos los casos nuevos de demencia cada año son pacientes con alzhéimer. También hay diferencias de incidencia dependiendo del sexo, ya que se aprecia un riesgo mayor de padecer la enfermedad en las mujeres, en particular entre la población mayor de 85 años.[36][37]
La prevalencia es el porcentaje de una población dada con una enfermedad. La edad avanzada es el principal factor de riesgo para sufrir alzhéimer: mayor frecuencia a mayor edad. La Organización Mundial de la Salud estimó que, en 2005, el 0,379 % de las personas a nivel mundial tenían demencia, y que la prevalencia aumentaría a un 0,441 % en 2015 y a un 0,556 % en 2030.[38]
Por otro lado, la Alzheimer's Disease International ha estimado para el año 2010 una prevalencia de demencia del 4,7 % a nivel mundial para personas de 60 años de edad o más,[39] representando por cierto cifras al alza respecto a varios estudios publicados con anterioridad (10 % superiores a las estimadas para The Lancet en 2005).[40] Otro estudio estimó que en el año 2006 un 0,4 % de la población mundial (entre 0,17-0,89 %; valor absoluto: aproximadamente 26 600 000, con un rango entre 11 400 000 y 59 400 000) se vio afectada por alzhéimer, y que la prevalencia triplicaría para el año 2050.[41]
En 2015 la Primera Conferencia Ministerial de la OMS sobre la Acción Mundial contra la Demencia estimó en 47,5 millones el número de casos en el mundo.[42]
En los Estados Unidos, la prevalencia del alzhéimer fue de un 1,6 % en el año 2000, tanto en la población general como en la comprendida entre los 65 y 74 años de edad. Se apreció un aumento del 19 % en el grupo de los 75-84 años, y del 42 % en el de los mayores de 84 años.[43] Sin embargo, las tasas de prevalencia en las regiones menos desarrolladas del mundo son inferiores.[40]
En la población europea, un meta-análisis publicado en 2017 en la revista Neurología reveló que la prevalencia de alzhéimer entre los 65 y los 74 años de edad es de 0,97 %, aumentando a 7,66 % en el grupo de 75 a 84 años, y a 22,53 % en el grupo de más de 85 años. Este mismo estudio también corroboró que esta enfermedad neurodegenerativa aparece con mayor frecuencia en el sexo femenino, ya que las mujeres presentan una prevalencia de alzhéimer de 7,13 %, mientras que la prevalencia en los hombres es de 3,31 %.[44]
Etiología
editarLas causas del alzhéimer no han sido descubiertas completamente. Existen tres principales hipótesis para explicar el fenómeno: el déficit de la acetilcolina, la acumulación de amiloide o tau y los trastornos metabólicos.
Hipótesis colinérgica
editarLa más antigua de ellas, y en la que se basan la mayoría de los tratamientos disponibles en el presente, es la hipótesis colinérgica, la cual sugiere que el alzhéimer se debe a una reducción en la síntesis del neurotransmisor acetilcolina. Esta hipótesis no ha recibido un apoyo global por la razón de que los medicamentos que tratan una deficiencia colinérgica tienen reducida efectividad en la prevención o cura del alzhéimer, aunque se ha propuesto que los efectos de la acetilcolina dan inicio a una acumulación a tan grandes escalas que conlleva a la neuroinflamación generalizada que deja de ser tratable simplemente promoviendo la síntesis del neurotransmisor.[45][46]
Hipótesis de los trastornos metabólicos
editarAlgunas investigaciones recientes han relacionado la demencia,[47] incluyendo la enfermedad de Alzheimer,[48] con desórdenes metabólicos,[49] particularmente con la hiperglicemia y la resistencia a la insulina. La expresión de receptores de la insulina ha sido demostrada en las neuronas del sistema nervioso central, preferentemente en las del hipocampo. En estas neuronas, cuando la insulina se une a su receptor celular, se promueve la activación de cascadas de señalización intracelular que conducen al cambio de la expresión de los genes relacionados con los procesos de plasticidad sináptica y de las enzimas relacionadas con el despeje de la misma insulina y del beta-amiloide. Estas enzimas degradantes de insulina promueven la disminución de la toxicidad debida al amiloide en modelos animales.
En esencia, el alzhéimer puede ser considerado como una forma de diabetes de cerebro que tiene elementos tanto de resistencia a la insulina como de deficiencia de insulina. Para consolidar este concepto, se ha propuesto que el alzhéimer sea conocido como «diabetes tipo 3».[50]
El alzhéimer está asociado con una progresiva resistencia a la insulina cerebral en ausencia de DM2, de resistencia a la insulina periférica o de obesidad. Estudios post mortem demostraron que tanto molecular como bioquímicamente, así como la transducción de señales anormales en el alzhéimer eran virtualmente idénticas a lo que ocurría en la diabetes mellitus tipo I y II. Al administrar localmente drogas prodiabéticas, como la estreptozotocina, estas generan daño cognitivo, con deficiencia espacial y de memoria, así como neurodegeneración típica de alzhéimer, pero no generan diabetes mellitus.[51]
Exposición al aluminio
editarHa existido polémica en torno al papel que tiene el aluminio como factor de riesgo para la enfermedad de Alzheimer, y esta hipótesis ha sido abandonada por numerosos científicos en 2014.[52]
Algunos estudios han mostrado en 2016 que el aluminio se asocia a varios procesos neurofisiológicos que provocan la característica degeneración del alzhéimer.[53][54] Sin embargo, algunas personas que han estado crónicamente expuestas al aluminio, a través de alimentos o agua, no han mostrado ningún síntoma de la enfermedad. La probable explicación es que su intestino mantiene la función de barrera protectora, que evita el paso de sustancias tóxicas a la sangre y las consecuentes reacciones.[53]
Consumo de gluten
editarSe han documentado casos de asociación de demencia por alzhéimer con el consumo de gluten y la mejoría con el seguimiento de una dieta sin gluten.[21]
Hipótesis de las proteínas β-amiloide y tau
editarOtra hipótesis propuesta en 1991[55][56] se ha relacionado con el acúmulo anómalo de la proteína beta-amiloide (también llamada amiloide Aβ) y tau en el cerebro de los pacientes con alzhéimer.[57]
En una minoría de pacientes, la enfermedad se produce por la aparición de mutaciones en los genes PSEN1, PSEN2 y en el gen de la APP, localizado en el cromosoma 21. En este último caso la enfermedad aparece clásicamente en personas con el síndrome de Down (trisomía en el cromosoma 21), casi universalmente en los 40 años de vida y se transmite de padres a hijos (por lo que existen, habitualmente, antecedentes familiares de alzhéimer en los pacientes que desarrollan la enfermedad en edades precoces). Esa relación en el cromosoma 21, y la tan elevada frecuencia de aparición de la enfermedad en las trisomías de ese cromosoma, hacen que la teoría sea muy evidente.[58][59]
Para poder hablar de las presenilinas, debemos recordar que el alzhéimer está caracterizado por depósitos amiloideos en el cerebro (como apoya la segunda teoría de este artículo).[60]
Su componente principal es el péptido beta-amiloide de 42 aminoácidos (βA42), en cuyo proceso de producción es fundamental la participación de la γ-secretasa, la cual depende a su vez de las presenilinas (PSEN).[61]
De esta manera se instituye un nuevo grupo de moléculas implicadas en la génesis de la enfermedad de Alzheimer y fundamental para su comprensión. Además son de notable actualidad debido a que su descubrimiento es relativamente reciente y a las posibilidades que ofrecen como dianas terapéuticas.[62]
Para comprender qué son estas moléculas, debemos señalar que se trata de un grupo de sustancias peptídicas producidas principalmente en el cerebro.[62][63]
Hay dos tipos, PSEN 1 y PSEN 2, con una estructura similar. La función principal que desempeñan ambas PSEN consiste en el procesamiento proteolítico de numerosas proteínas de membrana de tipo 1, entre ellas la APP, formando parte de la γ secretasa; de ahí la importancia de las PSEN en la enfermedad de Alzheimer, ya que a través de la regulación de la γ secretasa determinan la forma de Aβ que se genera y por tanto su acumulación en el tejido cerebral.[61][64][65]
El alzhéimer de inicio temprano se ha relacionado con mutaciones en el cromosoma 21, que contiene el gen de la PPA, y los cromosomas 14 y 1, que codifican para PSEN1 y PSEN2, respectivamente. Estas mutaciones tienen como resultado, entre otros efectos, el aumento de la concentración de βA, mientras que el alzhéimer de inicio tardío se relaciona con mutaciones en el gen de la apolipoproteina E.[60][66]
El gen que codifica la PSEN1, del que se conocen 177 mutaciones distintas, es el responsable de la aparición del alzhéimer de inicio tan temprano como a los 23 años de edad. La mutación de la PSEN2 es la causante de menos del 1 % de los casos de alzhéimer autosómicos dominantes, influyendo más en estos portadores los factores ambientales.[67][68][69][70]
La revisión de diferentes artículos nos ha conducido a la conclusión de que existe una relación entre las PSEN y el alzhéimer. Actualmente el avance en las técnicas de secuenciación del genoma ha aportado gran cantidad de información sobre las PSEN, su función, la localización en nuestros genes, la implicación de estas en la formación de βA, etc. No obstante, hoy en día aún se observan lagunas en cuanto a los mecanismos moleculares en los que están implicadas las PSEN.
Otro gran factor de riesgo genético es la presencia del gen de la APOE4 (apolipoproteína relacionada con la hiperlipoproteinemia y la hipercolesterolemia familiar), el cual tiende a producir una acumulación amiloide en el cerebro antes de que aparezcan los primeros síntomas del alzhéimer. Por ende, la deposición del amiloide Aβ tiende a preceder la clínica del alzhéimer.[71]
Otras evidencias parten de los hallazgos en ratones genéticamente modificados, los cuales solo expresan un gen humano mutado, el de la APP, el cual invariablemente les causa el desarrollo de placas amiloides fibrilares.[72] Se descubrió una vacuna experimental que causaba la eliminación de estas placas pero no tenía efecto sobre la demencia.[73]
Los depósitos de las placas no tienen correlación con la pérdida neuronal.[74]
Esta observación apoya la hipótesis tau, la cual defiende que es esta proteína la que da inicio a la cascada de trastornos de la enfermedad de Alzheimer.[57] De acuerdo con este modelo, las tau hiperfosforiladas adoptan formas anómalas, distribuyéndose en largas hileras. Eventualmente forman ovillos de neurofibrillas dentro de los cuerpos de las células nerviosas.[75]
Cuando esto ocurre, los microtúbulos se desintegran, colapsando el sistema de transporte de la neurona. Ello puede dar inicio a las primeras disfunciones en la comunicación bioquímica entre una neurona y la otra y conllevar la muerte de estas células.[76]
Según un trabajo realizado por un equipo de científicos de la Universidad de Cambridge, en Reino Unido cuyos resultados se publican en Brain,[77] la proteína tau, causante de la muerte de las células nerviosas, se disemina por todo el cerebro en la enfermedad de Alzheimer y, por lo tanto, bloquear su propagación puede evitar que la enfermedad aflore.
Se considera que los síntomas del alzhéimer están causados por la acumulación en el cerebro de dos proteínas anómalas: la proteína beta amiloide y la proteína tau. Se cree que la beta amiloide se produce primero, fomentando la aparición y la diseminación de tau, y que es esta última la que destruye las células nerviosas, mermando la memoria y las funciones cognitivas.
Hasta hace unos años, solamente era posible observar la acumulación de estas proteínas mediante el examen de los cerebros de los pacientes post mortem. Sin embargo, los recientes desarrollos en la tomografía por emisión de positrones (PET) han permitido a los científicos comenzar a visualizar su acumulación en pacientes que están vivos.
La forma en que tau se presenta en todo el cerebro ha sido tema de especulación entre los científicos y se han propuesto tres hipótesis:
- Una hipótesis es que la tau anómala comienza en un punto y desde ahí se propaga a otras regiones, desencadenando una reacción en cadena. Esta idea es conocida como la «propagación transneuronal» y está respaldada por estudios en ratones. Cuando a un ratón se le inyecta una proteína tau humana anómala, esta se propaga rápidamente por todo el cerebro. Sin embargo, debe reconocerse que la cantidad de tau inyectada es mucho más alta que los niveles de tau observados en cerebros humanos y la proteína se propaga rápidamente por el cerebro de un ratón mientras se disemina lentamente por el cerebro humano.
- La hipótesis de la «vulnerabilidad metabólica», que considera que la proteína tau se produce localmente en las células nerviosas, pero que algunas regiones presentan mayores demandas metabólicas y, por lo tanto, son más vulnerables a la proteína. En estos casos, tau es un marcador de sufrimiento en las células.
- Y la última hipótesis, del «soporte trófico», también plantea que algunas regiones cerebrales son más vulnerables que otras, pero que esto tiene menos que ver con la demanda metabólica y más con la falta de nutrición en la región o con los patrones de expresión génica.
Los avances en el escaneo PET permiten actualmente hacer comparaciones entre estas hipótesis.
Los científicos analizaron las conexiones funcionales dentro de los cerebros de los pacientes de alzhéimer y las compararon con los niveles de tau. Sus hallazgos respaldan la idea de la propagación transneuronal, es decir, que tau comienza en un lugar y se propaga. Al mismo tiempo, encontraron que no se cumplían las predicciones para las últimas dos hipótesis.
Según Thomas Cope, el primer autor del estudio, la propagación transneuronal es la correcta, las áreas del cerebro que están más conectadas tienen la mayor acumulación de la proteína tau anómala y la transmitirán a sus conexiones. En la enfermedad de Alzheimer, la región cerebral más común para tau que aparece primero es la región de la memoria, en el área de la corteza entorrinal, que está al lado del hipocampo. Los primeros síntomas en el alzhéimer tienden a ser problemas de memoria y el estudio de Cope sugiere que la tau luego se propaga por el cerebro, infectando y destruyendo las células nerviosas a medida que avanza, lo que hace que los síntomas del paciente empeoren progresivamente.
Esta confirmación es importante porque indica que se puede ralentizar o incluso detener la progresión de la enfermedad de Alzheimer mediante el desarrollo de fármacos para evitar que tau se mueva a lo largo de las neuronas.
Hipótesis microbiológicas
editarExisten numerosas hipótesis que han sido manejadas a lo largo de la historia sobre la patogenia del alzhéimer y que no han sido confirmadas por estudios independientes posteriores.
En 2013, un equipo de investigación publicó un estudio estadístico[78] en el que halló correlación entre las infecciones fúngicas diseminadas y la enfermedad de Alzheimer. Esta hipótesis no ha sido confirmada.
En 2018, la hipótesis de un equipo plantea que la bacteria Porphyromonas gingivalis podría ser una de las causas de enfermedad de Alzheimer.[79] Esta hipótesis tampoco ha sido confirmada.
En 2020, un estudio correlacionó la disbiosis de la microbiota intestinal y la aparición de placas amiloides en el cerebro, típicas de la enfermedad de Alzheimer. Esta hipótesis no ha sido confirmada por estudios posteriores.[80]
Patogenia
editarLa enfermedad de Alzheimer se caracteriza por la pérdida de neuronas y sinapsis en la corteza cerebral y en ciertas regiones subcorticales. Esta pérdida resulta en una atrofia de las regiones afectadas, incluyendo una degeneración en el lóbulo temporal y parietal y partes de la corteza frontal y la circunvolución cingulada.[46]
Neuropatología
editarLa neurodegeneración en la enfermedad de Alzheimer se debe a dos procesos: en el primero interviene la proteína beta-amiloide, que se acumula formando placas en el exterior de las neuronas. A su vez, se produce una alteración en el comportamiento de la proteína tau, que comienza a formar fibras entretejidas dentro de la célula nerviosa, los llamados ovillos. Es probable que muchos individuos, en su vejez, desarrollen estas placas y ovillos como parte del proceso normal de envejecimiento. Sin embargo, los pacientes con alzhéimer tienen un mayor número en lugares específicos del cerebro, como el lóbulo temporal.[81]
Bioquímica
editarLa enfermedad de Alzheimer se ha definido como una enfermedad que desdobla proteínas o proteopatía, debido a la acumulación de proteínas Aβ y tau, anormalmente dobladas, en el cerebro.[82]
Las placas neuríticas están constituidas por pequeños péptidos de 39-43 aminoácidos de longitud, llamados beta-amiloides (abreviados A-beta o Aβ). El beta-amiloide es un fragmento que proviene de una proteína de mayor tamaño conocida como Proteína Precursora de Amiloide (APP, por sus siglas en inglés). Esta proteína es indispensable para el crecimiento de las neuronas, para su supervivencia y su reparación postdaño.[83][84]
En la enfermedad de Alzheimer, un proceso aún desconocido es el responsable de que la APP sea dividida en varios fragmentos de menor tamaño por enzimas que catalizan un proceso de proteólisis.[85]
Uno de estos fragmentos es la fibra del beta-amiloide, el cual se agrupa y deposita fuera de las neuronas en formaciones microscópicamente densas conocidas como placas seniles.[19][86]
La enfermedad de Alzheimer se considera, debido a la agregación anormal de la proteína tau, como una tauopatía. Las neuronas sanas están compuestas por citoesqueleto, una estructura intracelular de soporte, parcialmente hechas de microtúbulos. Estos microtúbulos actúan como rieles que guían los nutrientes y otras moléculas desde el cuerpo neuronal hasta los extremos de los axones y viceversa. Cada proteína tau estabiliza los microtúbulos cuando es fosforilada y por esa asociación se le denomina proteína asociada al microtúbulo. En el alzhéimer, la tau debido a cambios químicos que resultan en su hiperfosforilación, se une con otras hebras tau creando ovillos de neurofibrillas y de esta manera, desintegra el sistema de transporte de la neurona.[87]
Patología
editarNo se ha explicado por completo cómo la producción y agregación de los péptidos Aβ (beta Amiloides) desempeñan un papel rol en el alzhéimer.[88]
La fórmula tradicional de la hipótesis amiloide apunta a la acumulación de los péptidos Aβ como el evento principal que conlleva la degeneración neuronal. La acumulación de las fibras amiloides, que parece ser la forma anómala de la proteína responsable de la perturbación de la homeostasis del ion calcio intracelular, induce la muerte celular programada, llamada apoptosis.[89]
Se sabe también que la Aβ se acumula selectivamente en las mitocondrias de las células cerebrales afectadas en el alzhéimer y que es capaz de inhibir ciertas funciones enzimáticas, así como alterar la utilización de la glucosa por las neuronas.[90]
Varios mecanismos inflamatorios y la intervención de las citoquinas pueden también desempeñar un papel en la patología de la enfermedad de Alzheimer. La inflamación es el marcador general de daño en los tejidos en cualquier enfermedad y puede ser secundaria al daño producido por el alzhéimer, o bien, la expresión de una respuesta inmunológica.[91] Mediante datos de metilación en el ADN se estimó que los granulocitos podrían estar aumentados en pacientes con Alzheimer. Del mismo modo, se determinó que los pacientes diagnosticados con demencia poseían granulocitos aumentados pero en menor medida que el resto de pacientes con Alzheimer, pudiendo ser una causa o consecuencia de progresión en la enfermedad.[92]
Genética
editarLa gran mayoría de los pacientes de esta enfermedad, tienen o han tenido algún familiar con alzhéimer. También hay que decir que en una pequeña proporción de los pacientes, el alzhéimer es debido a una generación autosómica dominante, que hace que la enfermedad aparezca de forma temprana. En menos de un 10 % de los casos, el alzhéimer aparece antes de los 60 años de edad como consecuencia de mutaciones autosómicas dominantes, representando, apenas, un 0,01 % de todos los casos.[93][94][95]
Estas mutaciones se han descubierto en tres genes distintos: el gen de la proteína precursora de amiloide (la APP) y los genes de las presenilinas 1 y 2.[93] Si bien la forma de aparición temprana de la enfermedad de Alzheimer ocurre por mutaciones en tres genes básicos, la forma más común no se ha podido explicar con un modelo puramente genético. La presencia del gen de la apolipoproteína E es el factor de riesgo genético más importante para padecer alzhéimer, pero no permite explicar todos los casos de la enfermedad.[93]
En 1987, se descubrió la relación de la enfermedad de Alzheimer con el cromosoma 21. Esto fue importante porque la mayoría de los afectados por el «síndrome de Down», o trisomía del cromosoma 21, padecen lesiones neuropatológicas similares a las del alzhéimer. Dentro del cromosoma 21 encontramos el gen PPA. John Hardy y sus colaboradores en 1991 afirmaron que este gen estaba implicado en la enfermedad de Alzheimer en un reducido número de familias. Sin embargo, se considera que de entre 5-10 % de los familiares con la enfermedad precoz la padecen debido a una mutación de este gen.
Las investigaciones dentro de este gen se han centrado en el péptido Ab (todas las mutaciones se encuentran alrededor de este péptido).
Las mutaciones producían un aumento de las concentraciones del péptido Ab. Esto llevó a la formación de la hipótesis de «cascada amieloide» en los años 90.
La «cascada amieloide» consiste en que la gran producción de Ab llevaría a la formación de depósitos en formas de placas seniles. Estas placas seniles serían nocivas para las células que producirían ovillos neurofibrilares, la muerte celular y la demencia.
Más tarde se vio en un grupo amplio de familias el ligamiento de la enfermedad de Alzheimer con el cromosoma 14. Pero esto llevó a una cadena de errores y con ello unas conclusiones erróneas.
Rudy Tanzi y Peter St George-Hyslop en 1995, mediante las técnicas de clonaje descubrieron otro gen S182 o Presenilin-1 (PS1). Este gen se encuentra entre los dominios 9 y 8 de transmembrana (con dos regiones hidrofílicas) y se le han encontrado más de 30 mutaciones.
Este gen interviene en procesos de apoptosis y es fundamental durante el desarrollo.
La mayoría de las mutaciones del gen Presenilin-1 (PS1) provocan un cambio en la estructura primaria. La PS1 y la enfermedad de Alzheimer no tienen una clara relación, pero hay que destacar que los pacientes tuvieron mutaciones que aumentan Ab en el plasma.
Poco más tarde se descubrió un nuevo gen que se denomina presenilina-2 (PS2) y también provoca el ascenso en la concentración de Ab, aunque las mutaciones observadas son de menor cantidad que los otros genes (PPA y PS1). La PS2 está formada por 8-9 dominios transmembrana.
La mayoría de las mutaciones en el gen de la APP y en los de las presenilinas, aumentan la producción de una pequeña proteína llamada beta-amiloide (Abeta 2), la cual es el principal componente de las placas seniles.[96]
Aunque la mayoría de los casos de alzhéimer no se deben a una herencia familiar, ciertos genes actúan como factores de riesgo. Un ejemplo es la transmisión familiar del alelo e4 del gen de la apolipoproteína E. Este gen se considera un factor de riesgo para la aparición de alzhéimer esporádico en fases tardías, produciendo un 50 % de los casos de alzhéimer.[97] Además de este, alrededor de 400 genes han sido también investigados por su relación con el alzhéimer esporádico en fase tardía.[93] Por ejemplo, en un meta-análisis GWAS publicado en 2019 los autores identificaron 16 genes asociados de forma significativa al alzhéimer de inicio tardío y observaron que 7 de ellos (HLA-DRA, HLA-DRB1, PTK2B, CLU, MS4A3, SCIMP y RABEP1) no estaban localizados en el locus APOE, por lo que podrían ser también de gran interés para futuras líneas de investigación en esta enfermedad neurodegenerativa.[98] En un nuevo estudio de asociación del genoma completo (GWAS) publicado en 2021, se identificaron 7 nuevos genes (TNIP1, HAVCR2, TMEM106B, GRN, LILRA5, AGRN, NTN5) asociados al desarrollo de Alzheimer esporádico o de inicio tardío (LOAD) no localizados en el locus de APOE. 5 de estos 7 genes (TNIP1, HAVCR2, LILRA5, AGRN, TNT5) no se habían asociado previamente con el desarrollo de enfermedades neurodegenerativas. Estas nuevas posibles variantes causales de la enfermedad resultan de gran interés puesto que podrían utilizarse como nuevas dianas terapéuticas para el tratamiento del Alzheimer.[99]
Epigenética
editarAdemás de estas variantes genéticas identificadas mediante GWAS, se ha comprobado que las modificaciones epigenéticas también tienen un papel importante. En un estudio conjunto de asociación del metiloma completo (MWAS), en el que además se incluyeron cohortes de Parkinson y ELA, se determinó que había 12 posiciones diferencialmente metiladas en pacientes con estas enfermedades neurodegenerativas. La asociación más fuerte se produjo en la región promotora del gen FKBP5.[92] La expresión de este gen aumenta progresivamente con la edad y este aumento se correlacionaría con un incremento en la patología de tau.[100]
Así pues, los genetistas coinciden en que hay más genes que actúan como factores de riesgo, aunque también afirman que existen otros que tienen ciertos efectos protectores que conllevan a retrasar la edad de la aparición del alzhéimer.[93] Un ejemplo es la alteración en el gen de la reelina, que contribuye a aumentar el riesgo de aparición del alzhéimer en mujeres.[101]
Cuadro clínico
editarPredemencia
editarLos primeros síntomas se confunden, con frecuencia, con la vejez o estrés en el paciente.[12] Una evaluación neuropsicológica detallada es capaz de revelar leves dificultades cognitivas hasta 8 años antes de que la persona cumpla los criterios de diagnóstico.[102]
Estos signos precoces pueden tener un efecto sobre las actividades de la vida diaria.[103]
La deficiencia más notable es la pérdida de memoria, manifestada como la dificultad de recordar hechos recientemente aprendidos y una inhabilidad para adquirir nueva información.[104][105][106]
Dificultades leves en las funciones ejecutivas —atención, planificación, flexibilidad y razonamiento abstracto— o trastornos en la memoria semántica —el recordar el significado de las cosas y la interrelación entre los conceptos— pueden también ser síntomas en las fases iniciales del alzhéimer.[107][108]
Puede aparecer apatía, siendo uno de los síntomas neuropsiquiátricos persistentes a lo largo de la enfermedad.[109][110][111]
La fase preclínica de la enfermedad es denominada por algunos deterioro cognitivo leve,[112] pero aún existe debate sobre si el término corresponde a una entidad diagnóstica independiente o si, efectivamente, es la primera etapa de la enfermedad.[113]
Demencia inicial
editarLos síntomas en esta fase inicial van desde una simple e insignificante, pero a veces recurrente, pérdida de memoria (como la dificultad en orientarse uno mismo en lugares como calles al estar conduciendo el automóvil), hasta una constante y más persuasiva pérdida de la memoria conocida como memoria a corto plazo, presentando dificultades al interactuar en áreas de índole familiar como el vecindario donde el individuo habita.
Además de la recurrente pérdida de la memoria, una pequeña porción de los pacientes presenta dificultades para el lenguaje, el reconocimiento de las percepciones —agnosia— o en la ejecución de movimientos —apraxia— con mayor prominencia que los trastornos de la memoria.[114]
El alzhéimer no afecta las capacidades de la memoria de la misma forma. La memoria a largo plazo o memorias episódicas, así como la memoria semántica o de los hechos aprendidos y la memoria implícita, que es la memoria del cuerpo sobre cómo realizar las acciones (tales como sostener el tenedor para comer), se afectan en menor grado que las capacidades para aprender nuevos hechos o el crear nuevos recuerdos.[115][116]
Los problemas del lenguaje se caracterizan, principalmente, por reducción del vocabulario y disminución en la fluidez de las palabras, lo que conlleva a un empobrecimiento general del lenguaje hablado y escrito. El paciente con alzhéimer suele ser capaz de comunicar adecuadamente las ideas básicas.[117][118][119]
También aparece torpeza al realizar tareas motoras finas, tales como escribir, dibujar o vestirse, así como ciertas dificultades de coordinación y de planificación.[120]
El paciente mantiene su autonomía y solamente necesita supervisión cuando se trata de tareas complejas.[114]
En esta etapa es frecuente que la persona se desoriente en la calle y llegue a perderse, por lo que se recomienda tomar precauciones:
- Colocándole en la muñeca una pulsera con un número de teléfono de contacto.
- Avisando a los que conocen la situación, para que alerten a la familia en caso de encontrar deambulando al enfermo de alzhéimer.
- Usando un localizador GPS para personas con alzhéimer, con el que la familia siempre pueda saber dónde está. Existen localizadores por teleasistencia, en los que el cuidador debe llamar a una teleoperadora para saber la ubicación del enfermo que lleva el dispositivo, y localizadores directos, en los que el cuidador tiene un receptor con el que, pulsando un botón, ve en la pantalla un mapa y la posición exacta de la persona enferma.
Demencia moderada
editarConforme avanza la enfermedad, los pacientes pueden realizar tareas con cierta independencia (como usar el baño), pero requerirán asistencia para tareas más complejas (por ejemplo, ir al banco, pagar cuentas, etc.).[114] Paulatinamente, llega la pérdida de aptitudes, como las de reconocer objetos y personas. Además, pueden manifestarse cambios de conducta como, por ejemplo, arranques violentos incluso en personas que jamás han presentado este tipo de comportamiento.
Los problemas del lenguaje son cada vez más evidentes debido a una inhabilidad para recordar el vocabulario, lo que produce frecuentes sustituciones de palabras erróneas, una condición llamada parafasia. Las capacidades para leer y escribir empeoran progresivamente.[117][121]
Las secuencias motoras complejas se vuelven menos coordinadas, reduciendo la habilidad de la persona para hacer sus actividades rutinarias.[122]
Durante esta fase, también empeoran los trastornos de la memoria y el paciente empieza a dejar de reconocer a sus familiares y seres más cercanos.[123]
La memoria a largo plazo, que hasta ese momento permanecía intacta, se deteriora.[124]
En esta etapa se vuelven más notorios los cambios en la conducta. Las manifestaciones neuropsiquiátricas más comunes son las distracciones, el desvarío y los episodios de confusión al final del día (agravados por la fatiga, la poca luz o la oscuridad),[126] así como la irritabilidad y la labilidad emocional, que incluyen llantos o risas inapropiadas, agresión no premeditada e incluso resistencia a las personas a cargo de sus cuidados. En el 30% aproximadamente de los pacientes aparecen ilusiones en el reconocimiento de personas.[109][127] También puede aparecer la incontinencia urinaria.[128]
Estos síntomas estresan a los familiares y a las personas al cuidado del paciente y pueden verse reducidos si se le traslada a un centro de cuidados a largo plazo.[114][129]
Demencia avanzada
editarLa enfermedad trae deterioro de la masa muscular, perdiéndose la movilidad, lo que lleva al enfermo a un estado de encamamiento,[130] la incapacidad de alimentarse a sí mismo,[131] junto a la incontinencia, en aquellos casos en que la muerte no haya llegado aún por causas externas (infecciones por úlceras o neumonía, por ejemplo).[132][133]
El lenguaje se torna severamente desorganizado, llegándose a perder completamente.[117] A pesar de ello, se conserva la capacidad de recibir y enviar señales emocionales.[134]
Los pacientes no podrán realizar ni las tareas más sencillas por sí mismos y requerirán constante supervisión, quedando así completamente dependientes. Puede aún estar presente cierta agresividad, aunque es más frecuente ver extrema apatía y agotamiento.[114]
Síntomas y etapas
editarLos principales síntomas del alzhéimer son pérdidas de memoria a corto plazo, anosognosia (desconocimiento de la enfermedad), confabulaciones para tapar los huecos en el recuerdo de corto plazo (mienten o inventan cosas para rellenar el vacío o laguna en sus relatos), estado depresivo, problemas de atención, problemas con la orientación, cambios de personalidad, dificultades con el lenguaje y cambios de humor inexplicables, desorientación, entre otras. Esta enfermedad tiene tres etapas en las cuales presenta diferentes síntomas.
Etapa 1 o leve
editarCuando la enfermedad comienza a manifestarse, las personas con alzhéimer tienden a ser menos enérgicas y espontáneas. Muestran pérdidas mínimas de memoria y cambios de humor, y tardan en aprender y reaccionar. También se vuelven aislados, evitan a desconocidos o lugares nuevos y prefieren lo familiar. Exhiben confusión, tienen dificultades para la organización y planificación, se extravían fácilmente y ejercen un pobre juicio. Pueden tener dificultad para realizar tareas rutinarias y tienen problemas para comunicarse y comprender material escrito. Si la persona está empleada, la pérdida de memoria puede comenzar a afectar su rendimiento en el trabajo. Todo esto les causa frustración y enojo.
Etapa 2 o moderada
editarEn esta etapa, la persona con la enfermedad de Alzheimer todavía puede realizar tareas simples en forma autónoma, pero puede necesitar ayuda con actividades complicadas. Los enfermos olvidan acontecimientos recientes y su historia personal, y cada vez están más desorientados y desconectados de la realidad. Pueden confundir su pasado con el presente, y les cuesta comprender la situación actual, fecha y hora. También pueden tener problemas para reconocer a su círculo familiar. Aumentan los problemas del habla y comprensión; la lectura y escritura son más difíciles, y el individuo tiende a inventar palabras. Los afectados ya no pueden estar seguros por sí solos y suelen deambular. Conforme el paciente se hace más consciente de esta pérdida de control se torna depresivo, irritable e inquieto, o bien apático y aislado. Pueden experimentar trastornos del sueño y tienen dificultad para comer, vestirse y asearse.
Etapa 3 o grave
editarDurante esta fase final puede perder la capacidad para alimentarse por sí solo, hablar, reconocer personas y controlar funciones corporales. Su falta de memoria se agrava hasta ser casi completa, requiriendo asistencia constante. En un estado físico debilitado el paciente puede llegar a ser vulnerable a otras enfermedades y problemas respiratorios, sobre todo cuando debe estar confinado a la cama.
Diagnóstico
editarEl diagnóstico se basa primero en la historia y la observación clínicas, del profesional de la salud y la que es referida por los familiares, basada en las características neurológicas y psicológicas, así como en la ausencia de condiciones alternativas: un diagnóstico de exclusión.[135][136]
Luego durante unas semanas o meses se realizan pruebas de memoria y de funcionamiento o evaluación intelectual.[12] También se efectúan análisis de sangre y escáner para descartar diagnósticos alternativos.
No existe un test pre mortem para diagnosticar concluyentemente el alzhéimer. Se ha conseguido aproximar la certeza del diagnóstico a un 85 %, pero el definitivo debe hacerse con pruebas histológicas sobre tejido cerebral, generalmente obtenidas en la autopsia.[137]
Las pruebas de imagen cerebral —Tomografía axial computarizada (TAC), Resonancia magnética nuclear (RMN), tomografía por emisión de positrones (TEP) o la tomografía computarizada por emisión de fotón único— pueden mostrar diferentes signos de que existe una demencia, pero no especifican de qué demencia se trata.[138]
Por tanto, el diagnóstico de la enfermedad de Alzheimer se basa tanto en la presencia de ciertas características neurológicas y neuropsicológicas, como en la ausencia de un diagnóstico alternativo y se apoya en el escáner cerebral para detectar signos de demencia. Actualmente se están desarrollando nuevas técnicas de diagnóstico basadas en el procesamiento de señales electroencefalográficas.
Una vez identificada, la expectativa promedio de vida de los pacientes que viven con la enfermedad de Alzheimer es aproximadamente de 7 a 10 años, aunque se conocen casos en los que se llega antes a la etapa terminal, entre 4 y 5 años; también existe el otro extremo, donde pueden sobrevivir hasta 21 años.
Criterios de diagnóstico
editarLa Asociación del Alzhéimer es el organismo que ha establecido los criterios diagnósticos más comúnmente usados, registrados en los Criterios NINCDS-ADRDA del Alzhéimer.[139]
Estas pautas requieren que la presencia de un trastorno cognitivo y la sospecha de un síndrome demencial sean confirmadas con una evaluación neuropsicológica con vistas a categorizar el diagnóstico de alzhéimer en dos: posible o probable. La confirmación histológica, que incluye un examen microscópico del tejido cerebral, se precisa para el diagnóstico definitivo del alzhéimer. Estos criterios incluyen que la presencia de un trastorno cognitivo y la sospecha de un síndrome demencial sean confirmados por evaluaciones neuropsicológicas para distinguir entre un diagnóstico posible o uno probable de la enfermedad de Alzheimer. Se ha mostrado fiabilidad y validez estadística entre los criterios diagnósticos y la confirmación histológica definitiva.[140]
Son ocho los dominios cognitivos que con más frecuencia se dañan en el alzhéimer: la memoria, el lenguaje, la percepción, la atención, las habilidades constructivas y de orientación, la resolución de problemas y las capacidades funcionales. Estos parámetros son equivalentes a los evaluados en los Criterios NINCDS-ADRDA publicados por la Asociación Americana de Psiquiatría.[141][142]
Herramientas de diagnóstico
editarLas evaluaciones neuropsicológicas, inclusive el examen minimental, son ampliamente usadas para evaluar los trastornos cognitivos necesarios para el diagnóstico de EA. Otra serie de exámenes más comprensivos son necesarios para una mayor fiabilidad en los resultados, especialmente en las fases iniciales de la enfermedad.[143][144]
El examen neurológico en los inicios del alzhéimer es crucial para el diagnóstico diferencial del alzhéimer y otras enfermedades.[12]
Las entrevistas a familiares también sirven para evaluar la enfermedad. Los cuidadores pueden proveer información y detalles importantes sobre las habilidades rutinarias, así como la disminución en el tiempo de la función mental del paciente.[145]
El punto de vista de la persona a cargo del paciente es de especial importancia debido a que el paciente, por lo general, no está al tanto de sus propias deficiencias.[146]
Muchas veces, los familiares tienen desafíos en la detección de los síntomas y signos iniciales de la demencia y puede que no comuniquen la información de manera acertada al profesional de salud especializado.[147]
Los exámenes adicionales pueden proporcionar información de algunos elementos de la enfermedad y tienden a ser usados para descartar otros diagnósticos. Los exámenes de sangre pueden identificar otras causas de demencia que no sea el alzhéimer,[12] que pueden ser, en pocos casos, enfermedades reversibles.[148]
El examen psicológico para la depresión sería de valor, puesto que la depresión puede aparecer de manera concomitante con el alzhéimer, o bien ser la causa de los trastornos cognitivos.[149][150]
En los casos en que estén disponibles imágenes neurológicas especializadas, como la TEP o la tomografía de fotón único, pueden servir para confirmar el diagnóstico del alzhéimer junto con las evaluaciones del estatus mental del individuo.[151]
La capacidad de una tomografía computarizada por emisión de fotón único, para distinguir entre el alzhéimer y otras posibles causas en alguien que ya fue diagnosticado de demencia, parece ser superior a los intentos de diagnóstico mediante exámenes mentales y la historia del paciente.[152]
Una nueva técnica, conocida como PiB PET, se ha desarrollado para tomar imágenes, directamente y de forma clara, de los depósitos beta-amiloides in vivo, con el uso de un radiofármaco que se une selectivamente a los depósitos Aβ.[153]
Otro marcado objetivo reciente de la enfermedad de Alzheimer es el análisis del líquido cefalorraquídeo en busca de amiloides beta o proteínas tau.[154]
Ambos avances de la imagen médica han derivado en propuestas para cambiar los criterios diagnósticos.[12][139]
Marcadores para el diagnóstico
editarUno de los grandes objetivos del diagnóstico consistiría en la identificación de marcadores genéticos que permitiesen un diagnóstico de alzheimer en etapas iniciales o incluso antes del desarrollo de la propia enfermedad. Para la determinación de estos biomarcadores resultan útiles los estudios de asociación de genomas (también llamados GWAS). Un estudio publicado en 2019 ha observado mediante el genotipado de inversiones y análisis de asociación de genomas (GWAS) una inversión que podría relacionarse con riesgo de alzheimer, debido a que presenta desequilibrio de ligamiento con un SNP asociado a alzheimer.[155]
Tratamiento
editarActualmente la enfermedad de Alzheimer es incurable y terminal.
El tratamiento del alzhéimer se sustenta en dos pilares complementarios: el tratamiento no farmacológico y el tratamiento farmacológico. Las intervenciones psicosociales se usan conjuntamente con el tratamiento farmacológico.
Tratamientos farmacológicos
editarSe ha probado cierta eficacia de parte de los fármacos anticolinesterásicos, estos presentan una acción inhibidora de la colinesterasa, la enzima encargada de descomponer la acetilcolina (neurotransmisor que falta en la enfermedad de Alzheimer y que incide sustancialmente en la memoria y otras funciones cognitivas). Se han incorporado al tratamiento de la enfermedad nuevos fármacos que intervienen en la regulación de la neurotransmisión glutaminérgica. Con todo esto se ha mejorado un poco el comportamiento del enfermo en cuanto a la apatía, la iniciativa, la capacidad funcional y las alucinaciones, mejorando su calidad de vida. Sin embargo, es preciso remarcar que desde 2008 los registros de la mejoría obtenida con dichos fármacos es discreta, es decir, no se ha conseguido alterar el curso de la demencia subyacente.
El primer fármaco anticolinesterásico comercializado fue la tacrina, que hoy ha dejado de emplearse por su hepatotoxicidad. En 2008, en Europa y Norteamérica existían 4 fármacos disponibles, tres de ellos son inhibidores de la acetilcolinesterasa: donepezilo (comercializado como Aricept),[156] rivastigmina (comercializado como Exelon o Prometax)[157] incluyendo el parche de Exelon,[158] y galantamina (comercializado como Reminyl).[159]
Los tres presentan un perfil de eficacia similar con parecidos efectos secundarios. Estos últimos suelen ser alteraciones gastrointestinales, anorexia y trastornos del ritmo cardíaco. El cuarto medicamento es un antagonista de los receptores NMDA, la memantina. Ninguno de los cuatro se indica para retardar o detener el progreso de la enfermedad.
La reducción en la actividad de las neuronas colinérgicas es una de las características reconocidas de la enfermedad de Alzheimer.[160]
Los inhibidores de la acetilcolinesterasa se emplean para reducir la tasa de degradación de la acetilcolina, manteniendo así concentraciones adecuadas del neurotransmisor en el cerebro y deteniendo su pérdida causada por la muerte de las neuronas colinérgicas.[161]
Existen evidencias de que estos medicamentos tienen eficacia en las etapas leves y moderadas de la enfermedad,[162] aunque un poco menos de que sean útiles en la fase avanzada. Solamente el donepezilo se ha aprobado para este estado de la demencia.[163]
El uso de estos fármacos en los trastornos cognitivos leves no ha mostrado ser capaz de retardar la aparición del alzhéimer.[164]
Los efectos adversos más comunes incluyen náuseas y vómitos, ambos ligados al exceso colinérgico que de ellos deriva. Estos efectos aparecen entre el 10 y el 20 % aproximadamente de los tratados y tienen severidad leve a moderada. Entre los efectos secundarios menos frecuentes figuran calambres musculares, disminución de la frecuencia cardíaca, disminución del apetito y del peso corporal y un incremento en la producción de jugo gástrico.[165]
La memantina es un fármaco con un mecanismo de acción diferente,[166] que está indicado en las fases moderadas y avanzadas de la enfermedad. Su mecanismo de acción teórico se basa en antagonizar los receptores NMDA glutaminérgicos, usado en un principio como un agente antigripal.[167] El glutamato es un neurotransmisor excitatorio del sistema nervioso central. Al parecer, un exceso de estimulación glutaminérgica podría producir o inducir una serie de reacciones intraneuronales de carácter tóxico, causando la muerte celular por un proceso llamado excitotoxicidad, que consiste en una sobreestimulación de los receptores del glutamato. Esta excitotoxicidad no solo ocurre en pacientes con alzhéimer, sino también en otras enfermedades neurodegenerativas, como la enfermedad de Parkinson y la esclerosis múltiple.[167]
Los ensayos clínicos han demostrado una eficacia moderada en estos pacientes y un perfil de efectos secundarios aceptable. En 2005 se aprobó también su indicación en fases moderadas de la enfermedad, pero los efectos en las fases iniciales son aún desconocidos.[168]
Los efectos adversos de la memantina son infrecuentes y leves e incluyen alucinaciones, confusión, mareos, dolor de cabeza y fatiga.[169]
La combinación de memantina y donepezilo ha mostrado ser estadísticamente significativa pero marginalmente exitosa desde el punto de vista clínico.[170]
Además existen fármacos que mejoran algunos de los síntomas que produce esta enfermedad, entre los que se encuentran ansiolíticos, hipnóticos, neurolépticos y antidepresivos. Los fármacos antipsicóticos se indican para reducir la agresión y la psicosis en pacientes con alzhéimer que tienen problemas de conducta, pero se usan con moderación y no de forma rutinaria por razón de los serios efectos secundarios, como eventos cerebrovasculares, trastornos extrapiramidales y una reducción cognitiva.[171]
Tratamientos no farmacológicos
editarRetrasar el avance del alzhéimer
editarEl avance de la enfermedad puede ser más rápido o más lento en función del entorno de la persona con alzhéimer. No es una situación fácil y la familia tendrá que hacer grandes esfuerzos para ofrecerle a la persona con alzhéimer un entorno lo más favorable posible.
Aceleradores de la enfermedad. Se consideran aceleradores las siguientes situaciones: estrés familiar, cambios bruscos en las rutinas diarias, cambio a un domicilio nuevo y desconocido (como son las residencias de mayores).
Retardadores de la enfermedad. Se consideran retardadores las situaciones nombradas a continuación: ambiente familiar feliz, hacer ejercicio, socializar con los amigos u otras personas.
Intervención psicosocial
editarExisten ciertas evidencias de que la estimulación de las capacidades cognitivas ayuda a ralentizar la pérdida de estas funciones y habilidades. Esta estimulación consiste en trabajar aquellas áreas que aún conserva el paciente, de forma que el entrenamiento permita compensar las pérdidas que el paciente está sufriendo con la enfermedad.
Las intervenciones psicosociales se usan conjuntamente con el tratamiento farmacológico y se clasifican en abordajes orientados al comportamiento, las emociones, lo cognitivo y la estimulación. Las investigaciones sobre la efectividad de estas intervenciones aún no se encuentran disponibles y, de hecho, rara vez son específicas al alzhéimer, enfocándose en la demencia en general.[172]
Las intervenciones en el área del comportamiento intentan identificar y reducir los antecedentes y consecuencias de los problemas de la conducta. Este abordaje no ha mostrado éxito en mejorar el funcionamiento general del paciente, en especial en relación con su entorno,[173] pero ha podido ayudar a reducir ciertos problemas específicos del comportamiento, como la incontinencia urinaria.[174]
Siguen faltando datos de calidad sobre la efectividad de estas técnicas en otros problemas como las deambulaciones del paciente.[175][176]
Las intervenciones orientadas a las emociones comprenden la terapia de validación, la terapia de reminiscencia, la psicoterapia de apoyo, la integración sensorial (también denominada snoezelen) y la terapia de presencia estimuladora. La psicoterapia de apoyo ha tenido poco estudio científico formal, pero algunos especialistas la encuentran de utilidad en pacientes con trastornos leves.[172] La terapia de reminiscencia incluye la discusión de experiencias del pasado de manera individual o en grupo, muchas veces con la ayuda de fotografías, objetos del hogar, música y grabaciones u otras pertenencias del pasado. En esta terapia, igualmente, no hay muchos estudios de calidad sobre su efectividad, aunque puede resultar beneficiosa para la reestructuración cognitiva y el humor.[177]
El tratamiento con presencias estimuladas se basa en las teorías de la adherencia e implica escuchar voces grabadas de los familiares y seres más cercanos del paciente con alzhéimer. Las evidencias preliminares indican que dichas actividades reducen la ansiedad y los comportamientos desafiantes.[178][179]
Finalmente, la terapia de validación se basa en la aceptación de la realidad y la experiencia personal de otras personas, mientras que la integración sensorial se basa en ejercicios guiados que estimulan los sentidos. Aún no hay suficientes evidencias que apoyen el uso de estas terapias en pacientes con alzhéimer.[180][181]
La finalidad de las terapias cognitivo-conductuales, que incluyen la orientación y la rehabilitación cognitiva, es reducir las distorsiones cognitivas. La orientación hacia la realidad consiste en presentar información acerca de la época, el lugar o la persona, con el fin de aliviar su entendimiento acerca de sus alrededores y el lugar que ellos desempeñan en dichos sitios. Por otro lado, el entrenamiento cognitivo intenta mejorar las capacidades debilitadas al ejercitar las habilidades mentales del paciente. Ambos ejercicios han mostrado cierta efectividad en el mejoramiento de las capacidades cognitivas.[182][183]
Sin embargo, en algunos estudios, estos efectos fueron transitorios y en otros tenían un efecto negativo, pues añadían frustración al paciente, según los reportes.[172]
Los tratamientos orientados a la estimulación incluyen la arteterapia, la musicoterapia y las terapias asistidas por mascotas, el ejercicio físico y cualquier actividad recreativa. La estimulación tiene apoyo modesto al ser aplicada con la intención de mejorar la conducta, el humor y, en menor grado, el funcionamiento del paciente. Si bien son efectos importantes, el principal beneficio reportado entre las terapias de estimulación es el mejoramiento en las rutinas de la vida diaria del paciente.[172]
Cuidados
editarDebido a que el alzhéimer no tiene cura, con el tiempo el paciente cae en un estado de imposibilidad de autosuficiencia para cuidar de sí mismo, por lo que los cuidados por terceros son una medida vital para esa deficiencia y deben ser abordados cuidadosamente durante el curso de la enfermedad.
En las fases tempranas y moderadas, las modificaciones al ambiente donde vive el paciente y a su estilo de vida, pueden darle seguridad y reducir las cargas al cuidador.[184][185]
Algunos ejemplos de dichas modificaciones son la adherencia a rutinas simplificadas, como son la colocación de candados, el uso de una pulsera con el número de teléfono del cuidador (o soluciones más avanzadas como un localizador por GPS), el etiquetado de los objetos del hogar y el uso de utensilios modificados para la vida diaria.[172][186][187] Puede llegar el punto en que el paciente no sea capaz de alimentarse a sí mismo, de modo que debe empezar a ingerir sus alimentos en porciones más pequeñas o en dietas no sólidas con la ayuda de otras personas.[188] Cuando aparezca una dificultad para tragar, puede que sea indicado el uso de sondas gástricas. En tales casos, la efectividad médica y ética de tener que continuar alimentando al paciente son consideraciones importantes que deben tomar los cuidadores y los familiares del individuo.[189][190]
Las restricciones físicas rara vez están indicadas en cualquier fase de la enfermedad, aunque hay situaciones en que son necesarias para prevenir que el paciente con alzhéimer se dañe a sí mismo o a terceros.[172]
A medida que progresa la enfermedad, pueden aparecer distintas manifestaciones médicas, como, las enfermedades orales y dentales, úlceras de presión, desnutrición, problemas de higiene o infecciones respiratorias, urinarias, dermatológicas u oculares, entre otras. El manejo cuidadoso del paciente puede prevenir dichos problemas, pero si llegan a producirse, deben ser tratados bajo supervisión médica.[133][191] Durante las etapas finales de la enfermedad, el tratamiento se centra en mantener la calidad de vida hasta el fallecimiento.[192]
Tratamientos en fase de investigación
editarVacunas
editarIgualmente se están realizando experimentos con vacunas, basados en la idea de que si el sistema inmune puede ser entrenado para reconocer y atacar la placa beta-amiloide, podría revertirse la deposición de amiloide y parar la enfermedad. Los resultados iniciales en animales fueron prometedores. Sin embargo, cuando las primeras vacunas se probaron en seres humanos en 2002, se produjo inflamación cerebral, (meningoencefalitis), en una pequeña proporción de los participantes en el estudio, por lo que se detuvieron las pruebas. Se continuó estudiando a los participantes y se observó una mejora en la lentitud del progreso de la enfermedad. Recientemente se ha descubierto que la inflamación cerebral estaba producida por una serie de péptidos que se incluían con la vacuna AN-179, por lo que se está investigando la creación de una vacuna que no tenga dichos péptidos en su composición.
Se está probando una vacuna la ABvac40, como preventiva contra el alzhéimer desde el 2014. Su objetivo, es detener la producción de placas amiloides. La vacuna produciría anticuerpos encargados de eliminar los beta amiloides 40 y 42, que son los causantes de la neurodegeneración cerebral. Los ensayos de la vacuna se realizarán sobre un total de 24 personas: 16 pacientes diagnosticados y en estadio leve y ocho pacientes que reciben placebo. De acreditarse su inocuidad, la vacuna no estará en el mercado antes del 2018.[193]
Ultrasonido
editarEn marzo de 2015 se publicó en Science-Translational Medicine una aproximación completamente nueva al tema alzhéimer, que ha sido probada en ratones.[194]
La misma utiliza una manera particular de aplicar ultrasonido, dentro del tejido gris cerebral. Estas ondas de sonido resultaron capaces de abrir gentilmente la barrera hematoencefálica, que separa al cerebro de la sangre, y estimularon a las células de del Río Hortega o microglía. Estas células microgliales, una vez activadas, resultaron capaces de ir desintegrando y eliminar las aglutinaciones beta-amiloideas del alzhéimer. Los autores de la investigación informaron haber observado la restauración completa de las memorias en el 75 % de los ratones en los que ensayaron. Hallaron que los ratones así tratados desplegaron mejoras de la memoria en tres pruebas específicas. El equipo científico planea iniciar pruebas con animales de laboratorio superiores, como ovejas y simios, y espera ser autorizado a poner en marcha ensayos sobre seres humanos en el 2017.[195]
Por su parte, en las aproximaciones clínicas clásicas (farmacológicas), un estudio de 2014 afirmaba que en ratones el antidepresivo citalopram detuvo el crecimiento de las placas beta amiloides de la enfermedad de Alzheimer existentes y redujo la formación de nuevas placas en un 78 %. En un segundo experimento, los científicos administraron una dosis única de citalopram a 23 personas de entre 18 y 50 años de edad que no estaban cognitivamente deterioradas ni padecían depresión. Cuando obtuvieron muestras de líquido cefalorraquídeo a las 24 horas, observaron una reducción del 37 % en la producción de la proteína beta-amiloide.[196]
Si esta enfermedad está relacionada con la resistencia a la insulina, se presentan múltiples alternativas terapéuticas. Se está evaluando actualmente el uso de medicamentos empleados en el tratamiento de la diabetes. Estudios recientes muestran que la administración de insulina por vía intranasal mejora la función cognitiva de pacientes normales y con alzhéimer.[cita requerida] Una revisión sistemática de los ensayos clínicos hasta ahora desarrollados muestra resultados esperanzadores. Por otra parte, se ha propuesto el empleo de técnicas de inducción enzimática, con enzimas activas por la insulina.[cita requerida]
Células madre
editarOtra de las áreas de investigación es la medicina regenerativa. Se trata de inyectar en el cerebro del paciente células madre embrionarias o adultas para intentar detener el deterioro cognitivo. Ya se han hecho experimentos en humanos con resultados positivos.[cita requerida]
Marcapasos cerebral
editarLa estimulación cerebral intenta normalizar la actividad, con un dispositivo llamado neuroestimulador, similar a un marcapasos cardíaco. El dispositivo forma parte de un tratamiento llamado estimulación cerebral profunda (ECP), que involucra la liberación de impulsos eléctricos para regular la actividad cerebral. La investigación, llevada a cabo en la Escuela de Medicina Johns Hopkins, forma parte de un proyecto más amplio iniciado en Canadá, donde ya se implantó el marcapasos a seis pacientes con la enfermedad. El tratamiento logró que los pacientes —todos con formas moderadas de alzhéimer— mostraran un incremento en la actividad neuronal durante 13 meses.
La terapia de estimulación cerebral profunda se ha utilizado con personas que sufren la enfermedad de Parkinson. Ahora, la terapia podría ser una alternativa para revertir el deterioro cognitivo de las personas con alzhéimer.
Esta neurocirugía funcional busca reparar, modular o corregir un déficit en un sistema o red neurológica determinada. Lo que ocurre con el alzhéimer es que se altera la química cerebral y esto conduce a una actividad eléctrica anormal que puede expresarse en temblores, deterioro cognitivo o trastornos psiquiátricos. La aplicación para el alzhéimer todavía está en sus primeras etapas.[197]
Prevención
editarLos estudios globales sobre las diferentes medidas que se pueden tomar para prevenir o retardar la aparición de la enfermedad de Alzheimer han tenido resultados contradictorios y no se ha comprobado aún una relación causal entre los factores de riesgo y la enfermedad, ni se han atribuido a efectos secundarios específicos. Por el momento, no parece haber medidas definitivas para prevenir la aparición del alzhéimer.[198]
Varios estudios epidemiológicos han propuesto diversas relaciones entre ciertos factores modificables, tales como la dieta, los riesgos cardiovasculares, productos farmacéuticos o las actividades intelectuales entre otros, y la probabilidad de que en una población aparezca el alzhéimer. Por ahora se necesitan más investigaciones y ensayos clínicos para comprobar si estos factores ayudan a prevenirla.[199][200]
La dieta mediterránea se ha asociado con un menor riesgo de desarrollar la enfermedad de Alzheimer, por su papel en la prevención del desarrollo de enfermedades cardiovasculares y por sus efectos antiinflamatorios y antioxidantes.[201]
La curcumina del curry ha mostrado en estudios de 2001 y 2007 cierta eficacia en la prevención de daño cerebral en modelos de ratón.[202]
Similares propiedades se comunicaron en 2010 y 2012 para ensayos en ratones de la Withania somnifera (ashwagandha, ginseng indio).[203][204]
A pesar de que los riesgos cardiovasculares, como la hipercolesterolemia, hipertensión arterial, la diabetes y el tabaquismo, están asociados a un mayor riesgo de desarrollo y progresión del alzhéimer,[205][206] las estatinas, que son medicamentos que disminuyen la concentración de colesterol en el plasma sanguíneo, no han sido efectivas en la prevención o mejoramiento del alzhéimer.[207][208] Sin embargo, en algunos individuos, el uso a largo plazo de los antiinflamatorios no esteroideos (AINEs) está vinculado con una reducción de la probabilidad de padecerla.[209]
Otros fármacos y terapias, como el reemplazo de hormonas en las mujeres, han dejado de ser aconsejadas como medidas preventivas del alzhéimer.[210][211]
Se incluye también un reporte en 2007 que concluyó la falta de evidencias significativas y la presencia de inconsistencias en el uso de ginkgo biloba para mejorar los trastornos cognitivos.[212]
Hay diferentes actividades intelectuales, como el jugar ajedrez, go, la lectura, el completar crucigramas o las interacciones sociales frecuentes, que parecen retardar la aparición y reducir la gravedad del alzhéimer.[213][214][215] El hablar varios idiomas también parece estar vinculado a la aparición tardía de la enfermedad.[216]
Otros estudios han sugerido un posible aumento en el riesgo de la aparición del alzhéimer con la exposición a campos magnéticos,[217][218] la ingestión de metales, en particular de aluminio,[219][220] o la exposición a ciertos solventes.[221] La calidad de algunos de estos estudios ha sido criticada,[222] y otros estudios han concluido que no hay una relación entre estos factores ambientales y la aparición del alzhéimer.[223][224][225][226]
Algunas asociaciones científicas actualmente trabajan en el campo médico y social recomendando y/o asesorando a las familias de los pacientes, propiciando a su vez la investigación, tales como Alzheimer's Association y Alzheimer Argentina.[227][228]
Véase también
editarReferencias
editar- ↑ a b c d e f Kumar, Anil; Sidhu, Jaskirat; Goyal, Amandeep; Tsao, Jack W. (2021). Alzheimer Disease. StatPearls Publishing. PMID 29763097. Consultado el 25 de abril de 2021.
- ↑ Mendez, Mario F. (2012-11-XX). «Early-onset Alzheimer's Disease: Nonamnestic Subtypes and Type 2 AD». Archives of Medical Research (en inglés) 43 (8): 677-685. ISSN 0188-4409. doi:10.1016/j.arcmed.2012.11.009. Consultado el 25 de abril de 2021.
- ↑ Querfurth, Henry W.; LaFerla, Frank M. (28 de enero de 2010). «Alzheimer's Disease». New England Journal of Medicine (en inglés) 362 (4): 329-344. ISSN 0028-4793. doi:10.1056/NEJMra0909142. Consultado el 25 de abril de 2021.
- ↑ Según la entrada «alzhéimer», en el Diccionario panhispánico de dudas (2005) de las academias del español, aunque la pronunciación etimológica del apellido alemán es /álts jáimer/ (en AFI ˈʔalts hai mɐ), normalmente en español se pronuncia /alséimer/ o /alzéimer/.
- ↑
Se ha reportado que, por término medio, el alzhéimer tiene una sobrevida después del diagnóstico entre:
- 10 años: MUNOZ CHACON, Yalile. Demencia, el reto del presente siglo (artículo completo disponible en español). Acta méd. costarric. [en línea]. Junio de 2003, vol. 45, n.º 2 [consultado el 2 de enero de 2010], pp. 42-42. ISSN 0001-6004.
- 8-10 años: La enfermedad de Alzheimer en el año 2000 (artículo completo disponible en español). Rev Panam Salud Publica [en línea]. 2001, vol. 10, n.º 4 [consultado el 3 de enero de 2010], pp. 268-276. ISSN 1020-4989. doi: 10.1590/S1020-49892001001000012.
- 7-9 años: DONOSO S, Archibaldo y BEHRENS P, María Isabel. Variabilidad y variantes de la enfermedad de Alzheimer (artículo completo disponible en español). Rev. méd. Chile [en línea]. 2005, vol. 133, n.º 4 [consultado el 2 de enero de 2010], pp. 477-482. ISSN 0034-9887. doi: 10.4067/S0034-98872005000400013.
- 7-8 años: DONOSO S, Archibaldo. La enfermedad de Alzheimer (artículo completo disponible en español). Rev. chil. neuro-psiquiatr. v. 41, supl. 2, Santiago, noviembre de 2003. Último acceso: 2 de enero de 2010. ISSN 0717-9227. doi: 10.4067/S0717-92272003041200003.
- ↑ Brookmeyer, R., Gray, S., Kawas, C. (septiembre de 1998). «Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset». American Journal of Public Health 88 (9): 1337-1342. PMC 1509089. PMID 9736873. doi:10.2105/AJPH.88.9.1337.
- ↑ E. Kraepelin. Psychiatrie: Ein Lehrbuch für Studierende und Ärzte. II. Band (Barth Verlag, Leipzig, 1910)
- ↑ A. Alzheimer. Allg. Z. Psychiatr. 64,146 (1907)
- ↑ A. Alzheimer. Z. Ges. Neurol. Psychiat. 4, 356 (1911)
- ↑ a b c Berchtold, N. C., Cotman, C. W. (1998). «Evolution in the conceptualization of dementia and Alzheimer's disease: Greco-Roman period to the 1960s». Neurobiol. Aging 19 (3): 173-189. PMID 9661992. doi:10.1016/S0197-4580(98)00052-9.
- ↑ Belaustegui, Isabel. «Qué hacer ante la enfermedad de Alzheimer». Vida Potencial. Consultado el 21 de diciembre de 2022.
- ↑ a b c d e f g Waldemar, G., Dubois, B., Emre, M., et al. (enero de 2007). «Recommendations for the diagnosis and management of Alzheimer's disease and other disorders associated with dementia: EFNS guideline». Eur J Neurol 14 (1): e1-26. PMID 17222085. doi:10.1111/j.1468-1331.2006.01605.x.
- ↑ «Alzheimer's diagnosis of AD». Alzheimer's Research Trust. Consultado el 7 de agosto de 2018.
- ↑ Tabert, M. H., Liu, X., Doty, R. L., Serby, M., Zamora, D., Pelton, G. H., Marder, K., Albers, M. W., Stern, Y., Devanand, D. P. (2005). «A 10-item smell identification scale related to risk for Alzheimer's disease». Ann. Neurol. 58 (1): 155-160. PMID 15984022. doi:10.1002/ana.20533.
- ↑ «Understanding stages and symptoms of Alzheimer's disease». National Institute on Aging. 26 de octubre de 2007. Archivado desde el original el 16 de mayo de 2008. Consultado el 21 de febrero de 2008.
- ↑ Mölsä PK, Marttila RJ, Rinne UK (agosto de 1986). «Survival and cause of death in Alzheimer's disease and multi-infarct dementia». Acta Neurol Scand 74 (2): 103-107. PMID 3776457. doi:10.1111/j.1600-0404.1986.tb04634.x.
- ↑ Mölsä PK, Marttila RJ, Rinne UK (marzo de 1995). «Long-term survival and predictors of mortality in Alzheimer's disease and multi-infarct dementia». ActaNeurol Scand 91 (3): 159-164. PMID 7793228.
- ↑ Prion Prion protein and Alzheimer disease.
- ↑ a b Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J (junio de 2004). «The importance of neuritic plaques and tangles to the development and evolution of AD». Neurology 62 (11): 1984-1989. PMID 15184601.
- ↑ «Alzheimer's Disease Clinical Trials». US National Institutes of Health. Consultado el 18 de agosto de 2008.
- ↑ a b Makhlouf S, Messelmani M, Zaouali J, Mrissa R (15 de diciembre de 2017). «Cognitive impairment in celiac disease and non-celiac gluten sensitivity: review of literature on the main cognitive impairments, the imaging and the effect of gluten free diet». Acta Neurol Belg (Revisión). PMID 29247390. doi:10.1007/s13760-017-0870-z.
- ↑ «Can Alzheimer's disease be prevented» (pdf). National Institute on Aging. 29 de agosto de 2006. Archivado desde el original el 27 de febrero de 2008. Consultado el 29 de febrero de 2008.
- ↑ «The MetLife study of Alzheimer's disease: The caregiving experience» (PDF). MetLife Mature Market Institute. agosto de 2006. Archivado desde el original el 25 de junio de 2008. Consultado el 12 de febrero de 2008.
- ↑ Thompson CA, Spilsbury K, Hall J, Birks Y, Barnes C, Adamson J (2007). «Systematic review of information and support interventions for caregivers of people with dementia». BMC Geriatr 7: 18. PMC 1951962. PMID 17662119. doi:10.1186/1471-2318-7-18.
- ↑ Schneider J, Murray J, Banerjee S, Mann A (agosto de 1999). «EUROCARE: a cross-national study of co-resident spouse carers for people with Alzheimer's disease: I—Factors associated with carer burden». International Journal of Geriatric Psychiatry 14 (8): 651-661. PMID 10489656. doi:10.1002/(SICI)1099-1166(199908)14:8<651::AID-GPS992>3.0.CO;2-B. Consultado el 4 de julio de 2008.
- ↑ Murray J, Schneider J, Banerjee S, Mann A (agosto de 1999). «EUROCARE: a cross-national study of co-resident spouse carers for people with Alzheimer's disease: II—A qualitative analysis of the experience of caregiving». International Journal of Geriatric Psychiatry 14 (8): 662-667. PMID 10489657. doi:10.1002/(SICI)1099-1166(199908)14:8<662::AID-GPS993>3.0.CO;2-4.
- ↑ a b c
Alzheimer Alois (1907). «Über eine eigenartige Erikrankung der Hirnrinde [About a peculiar disease of the cerebral cortex]» [Acerca de una peculiar enfermedad de la corteza cerebral.]. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-Gerichtlich Medizin (en alemán) 64 (1-2): 146-148.
- Alzheimer Alois (1987). «About a peculiar disease of the cerebral cortex. By Alois Alzheimer, 1907 (Translated by L. Jarvik and H. Greenson).». Alzheimer Dis Assoc Disord 1 (1): 3-8. PMID 3331112.
- Maurer Ulrike, Maurer Konrad (2003). Alzheimer: the life of a physician and the career of a disease. Nueva York: Columbia University Press. p. 270. ISBN 0-231-11896-1.
- ↑ a b Bick et al., 1987, pags 1-3
- ↑ a b Mark F. Bear; Barry Connors; Michael paradiso, eds. (2008) [2007]. «Capítulo 2: Neuronas y glía» (Google libros). Escrito en Estados Unidos. Neurociencia. La exploración del cerebro [Neuroscience. Exploring the brain] (3a edición edición). México: Wolters Kluwer Health. pp. 23-49. ISBN 978-0-7817-6003-4. OCLC 262825102. Edición española: ISBN 978-84-96921-09-2. Consultado el 15 de marzo de 2012.
- ↑ Berrios G E (1990). «Alzheimer's disease: a conceptual history». Int. J. Ger. Psychiatry 5: 355-365. doi:10.1002/gps.930050603.
- ↑ Kraepelin Emil, Diefendorf A. Ross (translated by). (17 de enero de 2007). Clinical Psychiatry: A Textbook For Students And Physicians (Reprint).. Kessinger Publishing. p. 568. ISBN 1-4325-0833-4.
- ↑ Katzman Robert, Terry Robert D, Bick Katherine L (editors). (1978). Alzheimer's disease: senile dementia and related disorders. Nueva York: Raven Press. p. 595. ISBN 0-89004-225-X.
- ↑ Boller F, Forbes MM (junio de 1998). «History of dementia and dementia in history: an overview». J. Neurol. Sci. 158 (2): 125-133. PMID 9702682. doi:10.1016/S0022-510X(98)00128-2.
- ↑ giancarlo LA, Rocca WA, Schoenberg BS (septiembre de 1986). «Origin of the distinction between Alzheimer's disease and senile dementia: how history can clarify nosolog». Neurology 62 (41): 1497-1499. PMID 3531918.
- ↑ a b Bermejo-Pareja F, Benito-León J, Vega S, Medrano MJ, Román GC (enero de 2008). «Incidence and subtypes of dementia in three elderly populations of central Spain». J. Neurol. Sci. 264 (1-2): 63-72. PMID 17727890. doi:10.1016/j.jns.2007.07.021.
- ↑ a b Di Carlo A, Baldereschi M, Amaducci L, et al (febrero de 2002). «Incidence of dementia, Alzheimers disease, and vascular dementia in Italy. The ILSA Study». J Am Geriatr Soc 50 (1): 41-48. PMID 12028245.
- ↑ Andersen K, Launer LJ, Dewey ME, et al (diciembre de 1999). «Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. EURODEM Incidence Research Group». Neurology 53 (9): 1992-1997. PMID 10599770.
- ↑ World Health Organization (2006). Neurological Disorders: Public Health Challenges. Switzerland: World Health Organization. pp. 204-207. ISBN 978-92-4-156336-9.
- ↑ Alzheimer's Disease International (2010). World Alzheimer Report 2010: The Global Economic Impact of Dementia. Ireland: Alzheimer's Disease International. p. 56.
- ↑ a b Ferri, CP,; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; Jorm, A.; Mathers, C.; Menezes, PR.; Rimmer, E.; Scazufca, M.; Alzheimer's Disease International (diciembre de 2005). «Global prevalence of dementia: a Delphi consensus study» (PDF). The Lancet 366 (9503): 2112-2117. PMID 16360788. doi:10.1016/S0140-6736(05)67889-0. Archivado desde el original el 25 de junio de 2008. Consultado el 13 de junio de 2008.
- ↑ 2006 prevalence estimate:
- Brookmeyer R, Johnson E, Ziegler-Graham K, MH Arrighi (julio de 2007). «Forecasting the global burden of Alzheimer’s disease». Alzheimer's and Dementia 3 (3): 186-191. doi:10.1016/j.jalz.2007.04.381. Archivado desde el original el 7 de diciembre de 2008. Consultado el 18 de junio de 2008.
- ↑ «CALL FOR ACTION by the participants of the First WHO Ministerial Conference on Global Action Against Dementia (Geneva, 16-17 March 2015)» (en inglés). Ginebra: Organización Mundial de la Salud. 17 de marzo de 2015. Consultado el 26 de abril de 2015.
- ↑ Estimaciones para 2000, Estados Unidos:
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA (agosto de 2003). «Alzheimer disease in the US population: prevalence estimates using the 2000 census». Arch. Neurol. 60 (8): 1119-1122. PMID 12925369. doi:10.1001/archneur.60.8.1119.
- «Profiles of general demographic characteristics, 2000 census of population and housing, United States» (PDF). U.S. Census Bureau. 2001. Consultado el 27 de agosto de 2008.
- ↑ Niu, H.; Alvarez-Alvarez, I.; Guillén-Grima, F.; Aguinaga-Ontoso (2017). «Prevalencia e incidencia de la enfermedad de Alzheimer en Europa: metaanálisis». Neurología 32 (8): 523-532. doi:10.1016/j.nrl.2016.02.016. Consultado el 20 de enero de 2020.
- ↑ Shen ZX (2004). «Brain cholinesterases: II. The molecular and cellular basis of Alzheimer's disease». Med. Hypotheses 63 (2): 308-321. PMID 15236795. doi:10.1016/j.mehy.2004.02.031.
- ↑ a b Wenk GL (2003). «Neuropathologic changes in Alzheimer's disease». J Clin Psychiatry. 64 Suppl 9: 7-10. PMID 12934968.
- ↑ Stewart R, Liolitsa D. (1999). «Type 2 diabetes mellitus, cognitive impairment and dementia». Diabet Med (en inglés) 16 (2): 93-112. PMID 10229302. doi:10.1046/j.1464-549. Archivado desde el original el 18 de julio de 2014. Consultado el 21 de septiembre de 2021.
- ↑ Diabetes y Alzheimer:
- Biessels GJ, Kappelle LJ; Utrecht Diabetic Encephalopathy Study Group. [Increased risk of Alzheimer's disease in Type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology?] (en inglés). Biochem Soc Trans. 2005 Nov;33(Pt 5):1041-4. PMID 16246041.
- Pasquier F, Boulogne A, Leys D, Fontaine P. Diabetes mellitus and dementia. (en inglés). Diabetes Metab. 2006 Nov;32(5 Pt 1):403-14. PMID 17110895.
- Reed BR, Mungas DM, Kramer JH, Ellis W, Vinters HV, Zarow C, Jagust WJ, Chui HC. Profiles of neuropsychological impairment in autopsy-defined Alzheimer's disease and cerebrovascular disease. Brain. 2007 Mar;130(Pt 3):731-9. Epub 2007 Jan 31. PMID 17267522.
- ↑ Jagua A, Avila A. Insulina y enfermedad de Alzheimer: una diabetes tipo 3? Rev Fac Med Univ Nac Colomb 2007; 55(1):6-70
- ↑ Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. (2005). «Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease--is this type 3 diabetes?». J Alzheimers Dis 7 ((1)): 63-80.
- ↑ Suzanne M. de la Monte* (enero de 2012). «Brain Insulin Resistance and Deficiency as Therapeutic Targets in Alzheimer's Disease». Current Alzheimer Research.
- ↑ Lidsky TI (2014). «Is the Aluminum Hypothesis dead?». Journal of Occupational and Environmental Medicine 56 (5 Suppl): S73-79. PMC 4131942. PMID 24806729. doi:10.1097/jom.0000000000000063.
- ↑ a b Kandimalla R, Vallamkondu J, Corgiat EB, Gill KD (marzo de 2016). «Understanding Aspects of Aluminum Exposure in Alzheimer's Disease Development». Brain Pathol (Revisón) 26 (2): 139-154. PMID 26494454. doi:10.1111/bpa.12333.
- ↑ FERREIRA, Pricilla Costa, PIAI, Kamila de Almeida, TAKAYANAGUI, Angela Maria Magosso et al. Aluminum as a risk factor for Alzheimer's disease. Rev. Latino-Am. Enfermagem [online]. 2008, vol. 16, n.º 1 [citado 2008-09-10], pp. 151-157. Disponible en la World Wide Web: [1]. ISSN 0104-1169. doi: 10.1590/S0104-11692008000100023
- ↑ Hardy J, Allsop D (octubre de 1991). «Amyloid deposition as the central event in the aetiology of Alzheimer's disease». Trends Pharmacol. Sci. 12 (10): 383-388. PMID 1763432.
- ↑ Selkoe, Dennis J; Hardy, John (1 de junio de 2016). «The amyloid hypothesis of Alzheimer's disease at 25 years». EMBO Molecular Medicine 8 (6): 595-608. ISSN 1757-4676. PMC 4888851. PMID 27025652. doi:10.15252/emmm.201606210. Consultado el 23 de julio de 2021.
- ↑ a b Mudher A, Lovestone S (enero de 2002). «Alzheimer's disease-do tauists and baptists finally shake hands?». Trends Neurosci. 25 (1): 22-26. PMID 11801334.
- ↑ Nistor M, Don M, Parekh M, et al (octubre de 2007). «Alpha- and beta-secretase activity as a function of age and beta-amyloid in Down syndrome and normal brain». Neurobiol. Aging 28 (10): 1493-1506. PMID 16904243. doi:10.1016/j.neurobiolaging.2006.06.023.
- ↑ Lott IT, Head E (marzo de 2005). «Alzheimer disease and Down syndrome: factors in pathogenesis». Neurobiol. Aging 26 (3): 383-389. PMID 15639317. doi:10.1016/j.neurobiolaging.2004.08.005.
- ↑ a b Small D, Klaver D, Foa L. (2010). «Presenilins and the γ-secretase: still a complex problem.». Mol Brain.: 1-6.
- ↑ a b García S, Vázquez R, Dávalos E, Castillo J, Martínez S, Ortiz A. (2009). «Enfermedad de Alzheimer: una panorámica de su primera descripción hacia una perspectiva molecular.». Med Int Mex. 25: 300-312.
- ↑ a b Vetrivel K, Zhang Y, Xu H, Thinakaran G. (2006). «Physiological and pathologycal functions of presenilines.». J Mol Neurosci. 1: 1-12.
- ↑ Perez N, Menendez S, Rodriguez J. (2002). «inas, Apo E y enfermedad de Alzheimer.». Rev Cuba Inve Biom. 21: 262-269.
- ↑ Bekris L, Yu C, Bird T, Tsuang D. (2010). «Genetics of Alzheimer Disease.». J Geri Psyc Neur. 23: 213-227.
- ↑ Crews L, Masliah E. (2010). «Molecular mechanisms of neurodegeneration in Alzheimer’s disease.». Hum Mol Genet.: 2-9.
- ↑ Theuns J, Del-Favero J, Dermaut B, van Duijin C, Backhovens H, Van den Broeck M et al. (2009). «Genetic variability in the regulatory region of presenilin 1 associated with risk for Alzheimer’s disease and variable expression.». Hum Mol Genet. 9: 325-331.
- ↑ Tandun A, Fraser P (2002). «The presenilines.». Geno Biol 3: 1-9.
- ↑ Theuns J, Del-Favero J, Dermaut B, van Duijin C, Backhovens H, Van den Broeck M et al. (2000). «Genetic variability in the regulatory region of presenilin 1 associated with risk for Alzheimer’s disease and variable expression.». Hum Mol Genet 9: 325-331.
- ↑ Sherrington R, Froelich S, Sorbi S, Campion D, Chi H, Rogaeval EA et al (1996). «Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant.». Hum Mol Genet. 5: 985-988.
- ↑ Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, et al. (1995). «Candidate gene for the chromosome 1 familial Alzheimer’ disease locus.». Science. 269: 973-977.
- ↑ Polvikoski T, Sulkava R, Haltia M, et al (noviembre de 1995). «Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein». N. Engl. J. Med. 333 (19): 1242-1247. PMID 7566000.
- ↑ Ratones trasgénicos:
- Games D, Adams D, Alessandrini R, et al (febrero de 1995). «Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein». Nature 373 (6514): 523-527. PMID 7845465. doi:10.1038/373523a0.
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D (septiembre de 1996). «Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease». J. Neurosci. 16 (18): 5795-5811. PMID 8795633.
- Hsiao K, Chapman P, Nilsen S, et al (octubre de 1996). «Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice». Science (journal). 274 (5284): 99-102. PMID 8810256.
- ↑ Holmes C, Boche D, Wilkinson D, et al (julio de 2008). «Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial». Lancet 372 (9634): 216-223. PMID 18640458. doi:10.1016/S0140-6736(08)61075-2.
- ↑ Schmitz C, Rutten BP, Pielen A, et al (abril de 2004). «Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer's disease». Am. J. Pathol. 164 (4): 1495-1502. PMC 1615337. PMID 15039236.
- ↑ Goedert M, Spillantini MG, Crowther RA (julio de 1991). «Tau proteins and neurofibrillary degeneration». Brain Pathol. 1 (4): 279-286. PMID 1669718.
- ↑ Chun W, Johnson GV (2007). «The role of tau phosphorylation and cleavage in neuronal cell death». Front. Biosci. 12: 733-756. PMID 17127334.
- ↑ Cope, Thomas E.; Rittman, Timothy; Borchert, Robin J.; Jones, P. Simon; Vatansever, Deniz; Allinson, Kieren; Passamonti; Vazquez Rodriguez, Patricia; Bevan-Jones, W. Richard; O’Brien, John T.; Rowe, James B. (5 de enero de 2018). «Tau burden and the functional connectome in Alzheimer’s disease and progressive supranuclear palsy» [La carga de Tau y el conectoma funcional en la enfermedad de Alzheimer y la parálisis supranuclear progresiva]. Brain (en inglés) (awx347). ISSN 1460-2156. doi:10.1093/brain/awx347. Consultado el 9 de enero de 2018.
- ↑ Alonso R, Pisa D, Rábano A, Carrasco L. «Alzheimer's disease and disseminated mycoses.». Eur J Clin Microbiol Infect Dis. PMID 24452965.
- ↑ Dominy, Lynch, Ermini, Benedyk, Marczyk, Konradi, Nguyen, Haditsch, Raha, Griffin, Holsinger, Arastu-Kapur, Kaba, Lee, Ryder, Potempa, Mydel, Hellvard, Adamowicz, Hasturk, Walker, Reynolds, Faull, Curtis, Dragunow, Potempa (2019). «Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors». Science Advances 5 (1,): eaau3333.
- ↑ Marizzoni M., Cattaneo A., Mirabelli P., Festari C., Lopizzo N., Nicolosi V., Mombelli E., Mazzelli M., Luongo D., Naviglio D., Coppola L., Salvatore M., Frison G.B. (2020). «Short-Chain Fatty Acids and Lipopolysaccharide as Mediators Between Gut Dysbiosis and Amyloid Pathology in Alzheimer's Disease». Journal of Alzheimers Disease 78 (2). doi:10.3233/JAD-200306. Consultado el 17 de abril de 2021.
- ↑ Bouras C, Hof PR, Giannakopoulos P, Michel JP, Morrison JH (1994). «Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one-year autopsy population from a geriatric hospital». Cereb. Cortex 4 (2): 138-150. PMID 8038565.
- ↑ Hashimoto M, Rockenstein E, Crews L, Masliah E (2003). «Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases». Neuromolecular Med. 4 (1-2): 21-36. PMID 14528050. doi:10.1385/NMM:4:1-2:21.
- ↑ Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J (julio de 2006). «Synapse formation and function is modulated by the amyloid precursor protein». J. Neurosci. 26 (27): 7212-7221. PMID 16822978. doi:10.1523/JNEUROSCI.1450-06.2006.
- ↑ Turner PR, O'Connor K, Tate WP, Abraham WC (mayo de 2003). «Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory». Prog. Neurobiol. 70 (1): 1-32. PMID 12927332.
- ↑ Hooper NM (abril de 2005). «Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein». Biochem. Soc. Trans. 33 (Pt 2): 335-338. PMID 15787600. doi:10.1042/BST0330335.
- ↑ Ohnishi S, Takano K (marzo de 2004). «Amyloid fibrils from the viewpoint of protein folding». Cell. Mol. Life Sci. 61 (5): 511-524. PMID 15004691. doi:10.1007/s00018-003-3264-8.
- ↑ Hernández F, Avila J (septiembre de 2007). «Tauopathies». Cell. Mol. Life Sci. 64 (17): 2219-2233. PMID 17604998. doi:10.1007/s00018-007-7220-x.
- ↑ Van Broeck B, Van Broeckhoven C, Kumar-Singh S (2007). «Current insights into molecular mechanisms of Alzheimer disease and their implications for therapeutic approaches». Neurodegener Dis 4 (5): 349-365. PMID 17622778. doi:10.1159/000105156.
- ↑ Yankner BA, Duffy LK, Kirschner DA (octubre de 1990). «Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides». Science (journal). 250 (4978): 279-282. PMID 2218531.
- ↑ Chen X, Yan SD (diciembre de 2006). «Mitochondrial Abeta: a potential cause of metabolic dysfunction in Alzheimer's disease». IUBMB Life 58 (12): 686-694. PMID 17424907. doi:10.1080/15216540601047767.
- ↑ Greig NH, Mattson MP, Perry T, et al (diciembre de 2004). «New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors,». Ann. N. Y. Acad. Sci. 1035: 290-315. PMID 15681814. doi:10.1196/annals.1332.018.
- ↑ a b Nabais, Marta F.; Laws, Simon M.; Lin, Tian; Vallerga, Costanza L.; Armstrong, Nicola J.; Blair, Ian P.; Kwok, John B.; Mather, Karen A. et al. (26 de marzo de 2021). «Meta-analysis of genome-wide DNA methylation identifies shared associations across neurodegenerative disorders». Genome Biology 22 (1): 90. ISSN 1474-760X. PMC 8004462. PMID 33771206. doi:10.1186/s13059-021-02275-5. Consultado el 26 de diciembre de 2021.
- ↑ a b c d e Waring SC, Rosenberg RN (marzo de 2008). «Genome-wide association studies in Alzheimer disease». Arch. Neurol. 65 (3): 329-334. PMID 18332245. doi:10.1001/archneur.65.3.329.
- ↑ Hoenicka J (marzo de 2006). «Genes in Alzheimer's disease». Rev Neurol 42 (5): 302-305. PMID 16538594.
- ↑ Campion D, Dumanchin C, Hannequin D, et al (septiembre de 1999). «Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum». Am. J. Hum. Genet. 65 (3): 664-670. PMC 1377972. PMID 10441572. doi:10.1086/302553.
- ↑ Selkoe DJ (junio de 1999). «Translating cell biology into therapeutic advances in Alzheimer's disease». Nature 399 (6738 Suppl): A23-31. PMID 10392577.
- ↑ Strittmatter WJ, Saunders AM, Schmechel D, et al (marzo de 1993). «Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease». Proc. Natl. Acad. Sci. USA 90 (5): 1977-1981. PMC 46003. PMID 8446617.
- ↑ Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K. et al. (2019). «Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk». Nature Genetics 51 (3): 404-413. doi:10.1038/s41588-018-0311-9. Consultado el 16 de diciembre de 2019.
- ↑ Wightman, Douglas P.; Jansen, Iris E.; Savage, Jeanne E.; Shadrin, Alexey A.; Bahrami, Shahram; Holland, Dominic; Rongve, Arvid; Børte, Sigrid et al. (2021-09). «A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease». Nature Genetics (en inglés) 53 (9): 1276-1282. ISSN 1546-1718. doi:10.1038/s41588-021-00921-z. Consultado el 7 de diciembre de 2021.
- ↑ Blair, Laura J.; Nordhues, Bryce A.; Hill, Shannon E.; Scaglione, K. Matthew; O’Leary, John C.; Fontaine, Sarah N.; Breydo, Leonid; Zhang, Bo et al. (1 de octubre de 2013). «Accelerated neurodegeneration through chaperone-mediated oligomerization of tau». The Journal of Clinical Investigation (en inglés) 123 (10): 4158-4169. ISSN 0021-9738. doi:10.1172/JCI69003. Consultado el 26 de diciembre de 2021.
- ↑ Seripa D, Matera MG, Franceschi M, et al (julio de 2008). «The RELN locus in Alzheimer's disease». J Alzheimers Dis. 14 (3): 335-344. PMID 18599960.
- ↑ Preclinicos:
- Linn RT, Wolf PA, Bachman DL, et al (mayo de 1995). «The 'preclinical phase' of probable Alzheimer's disease. A 13-year prospective study of the Framingham cohort». Arch. Neurol. 52 (5): 485-490. PMID 7733843.
- Saxton J, Lopez OL, Ratcliff G, et al (diciembre de 2004). «Preclinical Alzheimer disease: neuropsychological test performance 1.5 to 8 years prior to onset». Neurology 63 (12): 2341-2347. PMID 15623697.
- Twamley EW, Ropacki SA, Bondi MW (septiembre de 2006). «Neuropsychological and neuroimaging changes in preclinical Alzheimer's disease». J Int Neuropsychol Soc 12 (5): 707-735. PMC 1621044. PMID 16961952. doi:10.1017/S1355617706060863.
- ↑ Perneczky R, Pohl C, Sorg C, Hartmann J, Komossa K, Alexopoulos P, Wagenpfeil S, Kurz A (2006). «Complex activities of daily living in mild cognitive impairment: conceptual and diagnostic issues». Age Ageing 35 (3): 240-245. PMID 16513677. doi:10.1093/ageing/afj054.
- ↑ MARTINS, Sergilaine Pereira and DAMASCENO, Benito Pereira. Prospective and retrospective memory in mild Alzheimer's disease. Arq. Neuro-Psiquiatr. [online]. 2008, vol. 66, no. 2b [Consultado el 10 de septiembre de 2008], pp. 318-322. Disponible en la World Wide Web: [2]. ISSN 0004-282X. doi: 10.1590/S0004-282X2008000300006
- ↑ Arnáiz E, Almkvist O (2003). «Neuropsychological features of mild cognitive impairment and preclinical Alzheimer's disease». Acta Neurol. Scand., Suppl. 179: 34-41. PMID 12603249. doi:10.1034/j.1600-0404.107.s179.7.x.
- ↑ Kazui H, Matsuda A, Hirono N, et al (2005). «Everyday memory impairment of patients with mild cognitive impairment». Dement Geriatr Cogn Disord 19 (5-6): 331-337. PMID 15785034. doi:10.1159/000084559. Consultado el 12 de junio de 2008.
- ↑ Rapp MA, Reischies FM (2005). «Attention and executive control predict Alzheimer disease in late life: results from the Berlin Aging Study (BASE).». American Journal of Geriatric Psychiatry 13 (2): 134-141. PMID 15703322. doi:10.1176/appi.ajgp.13.2.134.
- ↑ Spaan PE, Raaijmakers JG, Jonker C (2003). «Alzheimer's disease versus normal ageing: a review of the efficiency of clinical and experimental memory measures». Journal of Clinical Experimental Neuropsychology 25 (2): 216-233. PMID 12754679.
- ↑ a b Craig D, Mirakhur A, Hart DJ, McIlroy SP, Passmore AP (2005). «A cross-sectional study of neuropsychiatric symptoms in 435 patients with Alzheimer's disease». American Journal of Geriatric Psychiatry 13 (6): 460-468. PMID 15956265. doi:10.1176/appi.ajgp.13.6.460.
- ↑ Robert PH, Berr C, Volteau M, Bertogliati C, Benoit M, Sarazin M, Legrain S, Dubois B (2006). «Apathy in patients with mild cognitive impairment and the risk of developing dementia of Alzheimer's disease: a one-year follow-up study». Clin Neurol Neurosurg 108 (8): 733-736. PMID 16567037. doi:10.1016/j.clineuro.2006.02.003.
- ↑ Palmer K, Berger AK, Monastero R, Winblad B, Bäckman L, Fratiglioni L (2007). «Predictors of progression from mild cognitive impairment to Alzheimer disease». Neurology 68 (19): 1596-1602. PMID 17485646. doi:10.1212/01.wnl.0000260968.92345.3f.
- ↑ Small BJ, Gagnon E, Robinson B (abril de 2007). «Early identification of cognitive deficits: preclinical Alzheimer's disease and mild cognitive impairment». Geriatrics 62 (4): 19-23. PMID 17408315.
- ↑ Petersen RC (febrero de 2007). «The current status of mild cognitive impairment--what do we tell our patients?». Nat Clin Pract Neurol 3 (2): 60-61. PMID 17279076. doi:10.1038/ncpneuro0402.
- ↑ a b c d e Förstl H, Kurz A (1999). «Clinical features of Alzheimer's disease». European Archives of Psychiatry and Clinical Neuroscience 249 (6): 288-290. PMID 10653284.
- ↑ Carlesimo GA, Oscar-Berman M (junio de 1992). «Memory deficits in Alzheimer's patients: a comprehensive review». Neuropsychol Rev 3 (2): 119-169. PMID 1300219.
- ↑ Jelicic M, Bonebakker AE, Bonke B (1995). «Implicit memory performance of patients with Alzheimer's disease: a brief review». International Psychogeriatrics 7 (3): 385-392. PMID 8821346. doi:10.1017/S1041610295002134.
- ↑ a b c Frank EM (septiembre de 1994). «Effect of Alzheimer's disease on communication function». J S C Med Assoc 90 (9): 417-423. PMID 7967534.
- ↑ Becker JT, Overman AA (2002). «The semantic memory deficit in Alzheimer's disease». Rev Neurol 35 (8): 777-783. PMID 12402233.
- ↑ Hodges JR, Patterson K (abril de 1995). «Is semantic memory consistently impaired early in the course of Alzheimer's disease? Neuroanatomical and diagnostic implications». Neuropsychologia 33 (4): 441-459. PMID 7617154.
- ↑ Benke T (diciembre de 1993). «Two forms of apraxia in Alzheimer's disease». Cortex 29 (4): 715-725. PMID 8124945.
- ↑ Forbes KE, Shanks MF, Venneri A (marzo de 2004). «The evolution of dysgraphia in Alzheimer's disease». Brain Res. Bull. 63 (1): 19-24. PMID 15121235. doi:10.1016/j.brainresbull.2003.11.005.
- ↑ Galasko D, Schmitt F, Thomas R, Jin S, Bennett D (2005). «Detailed assessment of activities of daily living in moderate to severe Alzheimer's disease». Journal of the International Neuropsychology Society 11 (4): 446-453. PMID 16209425.
- ↑ Galasko D, Schmitt F, Thomas R, Jin S, Bennett D (julio de 2005). «Detailed assessment of activities of daily living in moderate to severe Alzheimer's disease». J Int Neuropsychol Soc 11 (4): 446-453. PMID 16209425.
- ↑ Sartori G, Snitz BE, Sorcinelli L, Daum I (septiembre de 2004). «Remote memory in advanced Alzheimer's disease». Arch Clin Neuropsychol 19 (6): 779-789. PMID 15288331. doi:10.1016/j.acn.2003.09.007.
- ↑ por MedlinePlus (julio de 2008). «Úlcera de decúbito». Enciclopedia médica en español. Consultado el 10 de septiembre de 2008.
- ↑ Volicer L, Harper DG, Manning BC, Goldstein R, Satlin A (mayo de 2001). «Sundowning and circadian rhythms in Alzheimer's disease». Am J Psychiatry 158 (5): 704-711. PMID 11329390. Consultado el 27 de agosto de 2008.
- ↑ Síntomas neuropsiquiátricos:
- Scarmeas N, Brandt J, Blacker D, et al (diciembre de 2007). «Disruptive behavior as a predictor in Alzheimer disease». Arch. Neurol. 64 (12): 1755-1761. PMID 18071039. doi:10.1001/archneur.64.12.1755.
- Tatsch MF, Bottino CM, Azevedo D, et al (mayo de 2006). «Neuropsychiatric symptoms in Alzheimer disease and cognitively impaired, nondemented elderly from a community-based sample in Brazil: prevalence and relationship with dementia severity». Am J Geriatr Psychiatry 14 (5): 438-445. PMID 16670248. doi:10.1097/01.JGP.0000218218.47279.db.
- Volicer L, Bass EA, Luther SL (octubre de 2007). «Agitation and resistiveness to care are two separate behavioral syndromes of dementia». J Am Med Dir Assoc 8 (8): 527-532. PMID 17931577. doi:10.1016/j.jamda.2007.05.005.
- ↑ Honig LS, Mayeux R (junio de 2001). «Natural history of Alzheimer's disease». Aging (Milano). 13 (3): 171-182. PMID 11442300.
- ↑ Gold DP, Reis MF, Markiewicz D, Andres D (enero de 1995). «When home caregiving ends: a longitudinal study of outcomes for caregivers of relatives with dementia». J Am Geriatr Soc 43 (1): 10-16. PMID 7806732.
- ↑ Souren LE, Franssen EH, Reisberg B (junio de 1995). «Contractures and loss of function in patients with Alzheimer's disease». J Am Geriatr Soc 43 (6): 650-655. PMID 7775724.
- ↑ Berkhout AM, Cools HJ, van Houwelingen HC (septiembre de 1998). «The relationship between difficulties in feeding oneself and loss of weight in nursing-home patients with dementia». Age Ageing 27 (5): 637-641. PMID 12675103.
- ↑ Wada H, Nakajoh K, Satoh-Nakagawa T, et al (2001). «Risk factors of aspiration pneumonia in Alzheimer's disease patients». Gerontology 47 (5): 271-276. PMID 11490146.
- ↑ a b Gambassi G, Landi F, Lapane KL, Sgadari A, Mor V, Bernabei R (julio de 1999). «Predictors of mortality in patients with Alzheimer's disease living in nursing homes». J. Neurol. Neurosurg. Psychiatr. 67 (1): 59-65. PMC 1736445. PMID 10369823.
- ↑ Bär M, Kruse A, Re S (diciembre de 2003). «[Situations of emotional significance in residents suffering from dementia]». Z Gerontol Geriatr (en alemán) 36 (6): 454-462. PMID 14685735. doi:10.1007/s00391-003-0191-0.
- ↑ Mendez MF (2006). «The accurate diagnosis of early-onset dementia». International Journal of Psychiatry Medicine 36 (4): 401-412. PMID 17407994.
- ↑ Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J (noviembre de 2006). «Therapeutic approaches to Alzheimer's disease». Brain 129 (Pt 11): 2840-2855. PMID 17018549. doi:10.1093/brain/awl280.
- ↑ McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (julio de 1984). «Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease». Neurology 34 (7): 939-944. PMID 6610841.
- ↑ «Dementia: Quick reference guide» (PDF). Londres: (UK) National Institute for Health and Clinical Excellence. noviembre de 2006. ISBN 1-84629-312-X. Archivado desde el original el 27 de febrero de 2008. Consultado el 22 de febrero de 2008.
- ↑ a b Dubois B, Feldman HH, Jacova C, et al (agosto de 2007). «Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria». Lancet Neurol 6 (8): 734-746. PMID 17616482. doi:10.1016/S1474-4422(07)70178-3.
- ↑ Blacker D, Albert MS, Bassett SS, Go RC, Harrell LE, Folstein MF (diciembre de 1994). «Reliability and validity of NINCDS-ADRDA criteria for Alzheimer's disease. The National Institute of Mental Health Genetics Initiative». Arch. Neurol. 51 (12): 1198-1204. PMID 7986174.
- ↑ American Psychiatric Association (2000). Diagnostic and Statistical Manual of Mental disorders, 4th Edition Text Revision. Washington DC.
- ↑ Ito N (mayo de 1996). «[Clinical aspects of dementia]». Hokkaido Igaku Zasshi (en japonés) 71 (3): 315-320. PMID 8752526.
- ↑ Tombaugh TN, McIntyre NJ (septiembre de 1992). «The mini-mental state examination: a comprehensive review». J Am Geriatr Soc 40 (9): 922-935. PMID 1512391.
- ↑ Pasquier F (enero de 1999). «Early diagnosis of dementia: neuropsychology». J. Neurol. 246 (1): 6-15. PMID 9987708.
- ↑ Harvey PD, Moriarty PJ, Kleinman L, et al (2005). «The validation of a caregiver assessment of dementia: the Dementia Severity Scale». Alzheimer Dis Assoc Disord 19 (4): 186-194. PMID 16327345.
- ↑ Antoine C, Antoine P, Guermonprez P, Frigard B (2004). «[Awareness of deficits and anosognosia in Alzheimer's disease.]». Encephale (en francés) 30 (6): 570-577. PMID 15738860.
- ↑ Cruz VT, Pais J, Teixeira A, Nunes B (2004). «[The initial symptoms of Alzheimer disease: caregiver perception]». Acta Med Port (en portugués) 17 (6): 435-444. PMID 16197855.
- ↑ Clarfield AM (octubre de 2003). «The decreasing prevalence of reversible dementias: an updated meta-analysis». Arch. Intern. Med. 163 (18): 2219-2229. PMID 14557220. doi:10.1001/archinte.163.18.2219.
- ↑ Geldmacher DS, Whitehouse PJ (mayo de 1997). «Differential diagnosis of Alzheimer's disease». Neurology 48 (5 Suppl 6): S2-9. PMID 9153154.
- ↑ Potter GG, Steffens DC (mayo de 2007). «Contribution of depression to cognitive impairment and dementia in older adults». Neurologist 13 (3): 105-117. PMID 17495754. doi:10.1097/01.nrl.0000252947.15389.a9.
- ↑ Bonte FJ, Harris TS, Hynan LS, Bigio EH, White CL (julio de 2006). «Tc-99m HMPAO SPECT in the differential diagnosis of the dementias with histopathologic confirmation». Clin Nucl Med 31 (7): 376-378. PMID 16785801. doi:10.1097/01.rlu.0000222736.81365.63.
- ↑ Dougall NJ, Bruggink S, Ebmeier KP (2004). «Systematic review of the diagnostic accuracy of 99mTc-HMPAO-SPECT in dementia». Am J Geriatr Psychiatry 12 (6): 554-570. PMID 15545324. doi:10.1176/appi.ajgp.12.6.554.
- ↑ Sobre el PiB PET:
- Kemppainen NM, Aalto S, Karrasch M, et al (enero de 2008). «Cognitive reserve hypothesis: Pittsburgh Compound B and fluorodeoxyglucose positron emission tomography in relation to education in mild Alzheimer's disease». Ann. Neurol. 63 (1): 112-118. PMID 18023012. doi:10.1002/ana.21212.
- Ikonomovic MD, Klunk WE, Abrahamson EE, et al (junio de 2008). «Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease». Brain 131 (Pt 6): 1630-1645. PMC 2408940. PMID 18339640. doi:10.1093/brain/awn016.
- Jack CR, Lowe VJ, Senjem ML, et al (marzo de 2008). «11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment». Brain 131 (Pt 3): 665-680. PMID 18263627. doi:10.1093/brain/awm336.
- ↑ Marksteiner J, Hinterhuber H, Humpel C (junio de 2007). «Cerebrospinal fluid biomarkers for diagnosis of Alzheimer's disease: beta-amyloid(1-42), tau, phospho-tau-181 and total protein». Drugs Today 43 (6): 423-431. PMID 17612711. doi:10.1358/dot.2007.43.6.1067341.
- ↑ Giner-Delgado, C., Villatoro, S., Lerga-Jaso, J., Gayà-Vidal, M., Oliva, M., Castellano, D., ... & Olalde, I. (2019). Evolutionary and functional impact of common polymorphic inversions in the human Genome . Nature communications, 10(1), 1-14.
- ↑ «Donepezil». US National Library of Medicine (Medline Plus). 8 de enero de 2007. Consultado el 20 de marzo de 2008.
- ↑ «Rivastigmine». US National Library of Medicine (Medline Plus). 8 de enero de 2007. Consultado el 20 de marzo de 2008.
- ↑ «Rivastigmine Transdermal». US National Library of Medicine (Medline Plus). 8 de enero de 2007. Consultado el 20 de marzo de 2008.
- ↑ «Galantamine». US National Library of Medicine (Medline Plus). 8 de enero de 2007. Consultado el 20 de marzo de 2008.
- ↑ Geula C, Mesulam MM (1995). «Cholinesterases and the pathology of Alzheimer disease». Alzheimer Dis Assoc Disord. 9 Suppl 2: 23-28. PMID 8534419.
- ↑ Stahl SM (2000). «The new cholinesterase inhibitors for Alzheimer's disease, Part 2: illustrating their mechanisms of action». J Clin Psychiatry 61 (11): 813-814. PMID 11105732.
- ↑ Birks J (2006). «Cholinesterase inhibitors for Alzheimer's disease». Cochrane Database Syst Rev (1): CD005593. PMID 16437532. doi:10.1002/14651858.CD005593.
- ↑ Birks J, Harvey RJ (2006). «Donepezil for dementia due to Alzheimer's disease». Cochrane Database Syst Rev (1): CD001190. PMID 16437430. doi:10.1002/14651858.CD001190.pub2.
- ↑ Raschetti R, Albanese E, Vanacore N, Maggini M (2007). «Cholinesterase inhibitors in mild cognitive impairment: a systematic review of randomised trials». PLoS Med 4 (11): e338. PMID 18044984. doi:10.1371/journal.pmed.0040338.
- ↑ Información sobre los inhibidores de la acetilcolinesterasa:
- «Aricept Prescribing information» (PDF). Eisai and Pfizer. Archivado desde el original el 10 de septiembre de 2008. Consultado el 18 de agosto de 2008.
- «Razadyne ER U.S. Full Prescribing Information» (PDF). Ortho-McNeil Neurologics. Archivado desde el original el 28 de mayo de 2008. Consultado el 19 de febrero de 2008.
- «Exelon ER U.S. Prescribing Information» (PDF). Novartis Pharmaceuticals. Consultado el 19 de febrero de 2008.
- «Exelon U.S. Prescribing Information» (PDF). Novartis Pharmaceuticals. Archivado desde el original el 27 de febrero de 2008. Consultado el 21 de febrero de 2008.
- «Exelon Warning Letter». FDA. Archivado desde el original el 18 de enero de 2009.
- ↑ «Memantine». US National Library of Medicine (Medline). 4 de enero de 2004. Consultado el 22 de marzo de 2008.
- ↑ a b Lipton SA (2006). «Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond». Nat Rev Drug Discov 5 (2): 160-170. PMID 16424917. doi:10.1038/nrd1958.
- ↑ Areosa Sastre A, McShane R, Sherriff F (2004). «Memantine for dementia». Cochrane Database Syst Rev (4): CD003154. PMID 15495043. doi:10.1002/14651858.CD003154.pub2.
- ↑ «Namenda Prescribing Information» (PDF). Forest Pharmaceuticals. Archivado desde el original el 27 de febrero de 2008. Consultado el 19 de febrero de 2008.
- ↑ Raina P, Santaguida P, Ismaila A, et al (2008). «Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline». Annals of Internal Medicine 148 (5): 379-397. PMID 18316756.
- ↑ Uso de antipsicóticos:
- Ballard C, Waite J (2006). «The effectiveness of atypical antipsychotics for the treatment of aggression and psychosis in Alzheimer's disease». Cochrane Database Syst Rev (1): CD003476. PMID 16437455. doi:10.1002/14651858.CD003476.pub2.
- Ballard C, Lana MM, Theodoulou M, et al (2008). «A Randomised, Blinded, Placebo-Controlled Trial in Dementia Patients Continuing or Stopping Neuroleptics (The DART-AD Trial).». PLoS Med. 5 (4): e76. PMID 18384230. doi:10.1371/journal.pmed.0050076.
- Sink KM, Holden KF, Yaffe K (2005). «Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence». JAMA 293 (5): 596-608. PMID 15687315. doi:10.1001/jama.293.5.596.
- ↑ a b c d e f «Practice Guideline for the Treatment of Patients with Alzheimer's disease and Other Dementias» (PDF). American Psychiatric Association. octubre de 2007. doi:10.1176/appi.books.9780890423967.152139. Archivado desde el original el 25 de agosto de 2011. Consultado el 28 de diciembre de 2007.
- ↑ Bottino CM, Carvalho IA, Alvarez AM, et al (2005). «Cognitive rehabilitation combined with drug treatment in Alzheimer's disease patients: a pilot study». Clin Rehabil 19 (8): 861-869. PMID 16323385. doi:10.1191/0269215505cr911oa.
- ↑ Doody RS, Stevens JC, Beck C, et al (2001). «Practice parameter: management of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology». Neurology 56 (9): 1154-1166. PMID 11342679.
- ↑ Hermans DG, Htay UH, McShane R (2007). «Non-pharmacological interventions for wandering of people with dementia in the domestic setting». Cochrane Database Syst Rev (1): CD005994. PMID 17253573. doi:10.1002/14651858.CD005994.pub2.
- ↑ Robinson L, Hutchings D, Dickinson HO, et al (2007). «Effectiveness and acceptability of non-pharmacological interventions to reduce wandering in dementia: a systematic review». Int J Geriatr Psychiatry 22 (1): 9-22. PMID 17096455. doi:10.1002/gps.1643.
- ↑ Woods B, Spector A, Jones C, Orrell M, Davies S (2005). «Reminiscence therapy for dementia». Cochrane Database Syst Rev (2): CD001120. PMID 15846613. doi:10.1002/14651858.CD001120.pub2.
- ↑ Peak JS, Cheston RI (2002). «Using simulated presence therapy with people with dementia». Aging Ment Health 6 (1): 77-81. PMID 11827626. doi:10.1080/13607860120101095.
- ↑ Camberg L, Woods P, Ooi WL, et al (1999). «Evaluation of Simulated Presence: a personalised approach to enhance well-being in persons with Alzheimer's disease». J Am Geriatr Soc 47 (4): 446-452. PMID 10203120.
- ↑ Neal M, Briggs M (2003). «Validation therapy for dementia». Cochrane Database Syst Rev (3): CD001394. PMID 12917907. doi:10.1002/14651858.CD001394.
- ↑ Chung JC, Lai CK, Chung PM, French HP (2002). «Snoezelen for dementia». Cochrane Database Syst Rev (4): CD003152. PMID 12519587. doi:10.1002/14651858.CD003152.
- ↑ Spector A, Orrell M, Davies S, Woods B (2000). «Withdrawn: Reality orientation for dementia». Cochrane Database Syst Rev (3): CD001119. PMID 17636652. doi:10.1002/14651858.CD001119.pub2.
- ↑ Spector A, Thorgrimsen L, Woods B, et al (2003). «Efficacy of an evidence-based cognitive stimulation therapy programme for people with dementia: randomised controlled trial». Br J Psychiatry 183: 248-254. PMID 12948999. doi:10.1192/bjp.183.3.248.
- ↑ Gitlin LN, Corcoran M, Winter L, Boyce A, Hauck WW (febrero de 2001). «A randomized, controlled trial of a home environmental intervention: effect on efficacy and upset in caregivers and on daily function of persons with dementia». Gerontologist 41 (1): 4-14. PMID 11220813. Consultado el 15 de julio de 2008. (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).
- ↑ Gitlin LN, Hauck WW, Dennis MP, Winter L (marzo de 2005). «Maintenance of effects of the home environmental skill-building program for family caregivers and individuals with Alzheimer's disease and related disorders». J. Gerontol. A Biol. Sci. Med. Sci. 60 (3): 368-374. PMID 15860476.
- ↑ «Treating behavioral and psychiatric symptoms». Alzheimer's Association. 2006. Archivado desde el original el 25 de septiembre de 2006. Consultado el 25 de septiembre de 2006.
- ↑ Dunne TE, Neargarder SA, Cipolloni PB, Cronin-Golomb A (2004). «Visual contrast enhances food and liquid intake in advanced Alzheimer's disease». Clinical Nutrition 28 (6): 533-538. PMID 15297089. doi:10.1016/j.clnu.2003.09.015.
- ↑ Dudek, Susan G. (2007). Nutrition essentials for nursing practice. Hagerstown, Maryland: Lippincott Williams & Wilkins. p. 360. ISBN 0-7817-6651-6. Consultado el 19 de agosto de 2008.
- ↑ Dennehy C (2006). «Analysis of patients' rights: dementia and PEG insertion». Br J Nurs 15 (1): 18-20. PMID 16415742.
- ↑ Chernoff R (abril de 2006). «Tube feeding patients with dementia». Nutr Clin Pract 21 (2): 142-146. PMID 16556924.
- ↑ Problemas médicos:
- Head B (enero de 2003). «Palliative care for persons with dementia». Home Healthc Nurse 21 (1): 53-60; quiz 61. PMID 12544465.
- Friedlander AH, Norman DC, Mahler ME, Norman KM, Yagiela JA (septiembre de 2006). «Alzheimer's disease: psychopathology, medical management and dental implications». J Am Dent Assoc 137 (9): 1240-1251. PMID 16946428.
- Belmin J (2007). «Practical guidelines for the diagnosis and management of weight loss in Alzheimer's disease: a consensus from appropriateness ratings of a large expert panel». J Nutr Health Aging 11 (1): 33-37. PMID 17315078.
- McCurry SM, Gibbons LE, Logsdon RG, Vitiello M, Teri L (octubre de 2003). «Training caregivers to change the sleep hygiene practices of patients with dementia: the NITE-AD project». J Am Geriatr Soc 51 (10): 1455-1460. PMID 14511168.
- Perls TT, Herget M (diciembre de 1995). «Higher respiratory infection rates on an Alzheimer's special care unit and successful intervention». J Am Geriatr Soc 43 (12): 1341-1344. PMID 7490383.
- ↑ Shega JW, Levin A, Hougham GW, et al (abril de 2003). «Palliative Excellence in Alzheimer Care Efforts (PEACE): a program description». J Palliat Med 6 (2): 315-320. PMID 12854952. doi:10.1089/109662103764978641.
- ↑ «Comienzan los ensayos clínicos en humanos de la vacuna contra el alzhéimer.». europapress.es. 26 de febrero de 2014. Consultado el 27 de mayo de 2015.
- ↑ Gerhard Leinenga, Jürgen Götz (11 de marzo de 2015). «Scanning ultrasound removes amyloid-β and restores memory in an Alzheimer’s disease mouse model». Science Translational Medicine 7 (Issue 278): p. 278ra33. doi:10.1126/scitranslmed.aaa2512.
- ↑ Götz, Jürgen (16 de marzo de 2015). «Entrevista radial “16 March 2015 on Health Report”, ABC Radio (Australia)» (en inglés). Consultado el 23 de marzo de 2015.
- ↑ Cf. Yvette I. Sheline, Tim West, Kevin Yarasheski et al. "An Antidepressant Decreases CSF Aβ Production in Healthy Individuals and in Transgenic AD Mice", en Science Translational Medicine 14 de mayo de 2014: vol. 6, issue 236, p. 236re4 http://stm.sciencemag.org/content/6/236/236re4 Archivado el 16 de agosto de 2016 en Wayback Machine. y el más divulgativo artículo de ABC salud, http://www.abc.es/salud/noticias/20140515/abci-alzheimer-antidepresivo-nature-201405141801.html
- ↑
- ↑ No hay pruebas que recomienden medidas preventivas:
- Kawas CH (2006). «Medications and diet: protective factors for AD?». Alzheimer Dis Assoc Disord 20 (3 Suppl 2): S89-96. PMID 16917203.
- Luchsinger JA, Mayeux R (2004). «Dietary factors and Alzheimer's disease». Lancet Neurol 3 (10): 579-587. PMID 15380154. doi:10.1016/S1474-4422(04)00878-6.
- Luchsinger JA, Noble JM, Scarmeas N (2007). «Diet and Alzheimer's disease». Curr Neurol Neurosci Rep 7 (5): 366-372. PMID 17764625. doi:10.1007/s11910-007-0057-8.
- ↑ Szekely CA, Breitner JC, Zandi PP (2007). «Prevention of Alzheimer's disease». Int Rev Psychiatry 19 (6): 693-706. PMID 18092245. doi:10.1080/09540260701797944.
- ↑ «On the prevention and treatment of Alzheimer’s disease: Control the promoters and look beyond the brain». Medical Hypotheses (en inglés) 154: 110645. 1 de septiembre de 2021. ISSN 0306-9877. doi:10.1016/j.mehy.2021.110645. Consultado el 28 de julio de 2021.
- ↑ Aridi YS, Walker JL, Wright ORL (28 de junio de 2017). «The Association between the Mediterranean Dietary Pattern and Cognitive Health: A Systematic Review». Nutrients (Revisión) 9 (7): pii: E674. PMC 5537789. PMID 28657600. doi:10.3390/nu9070674.
- ↑ Curcumina en la dieta:
- Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ (2007). «Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model». Journal of Neurochemistry 102 (4): 1095-1104. PMID 17472706. doi:10.1111/j.1471-4159.2007.04613.x.
- Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM (2001). «The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse». Journal of Neuroscience 21 (21): 8370-8377. PMID 11606625.
- ↑ Sehgal N., Gupta A., Valli R.K., Joshi S.D., Mills J.T., Hamel E., Khanna P., Jain S.C., Thakur S.S. y Ravindranath V. (2012). «Withania somnifera reverses Alzheimer's disease pathology by enhancing low-density lipoprotein receptor-related protein in liver». Proc Natl Acad Sci U S A. (en inglés) 109 (9): 3510-3515. doi:10.1073/pnas.1112209109.
- ↑ Jayaprakasam B., Padmanabhan K. y Nair M.G. (2010). «Withanamides in Withania somnifera fruit protect PC-12 cells from beta-amyloid responsible for Alzheimer's disease». Phytother Res. (en inglés) 24 (6): 859-863. doi:10.1002/ptr.3033.
- ↑ Rosendorff C, Beeri MS, Silverman JM (2007). «Cardiovascular risk factors for Alzheimer's disease». Am J Geriatr Cardiol 16 (3): 143-149. PMID 17483665. doi:10.1111/j.1076-7460.2007.06696.x.
- ↑ Gustafson D, Rothenberg E, Blennow K, Steen B, Skoog I (2003). «An 18-year follow-up of overweight and risk of Alzheimer disease». Arch. Intern. Med. 163 (13): 1524-1528. PMID 12860573. doi:10.1001/archinte.163.13.1524.
- ↑ Reiss AB, Wirkowski E (2007). «Role of HMG-CoA reductase inhibitors in neurological disorders : progress to date». Drugs 67 (15): 2111-2120. PMID 17927279.
- ↑ Kuller LH (2007). «Statins and dementia». Current Atherosclerosis Reports 9 (2): 154-161. PMID 17877925. doi:10.1007/s11883-007-0012-9.
- ↑ Szekely CA, Breitner JC, Fitzpatrick AL, Rea TD, Psaty BM, Kuller LH, Zandi PP (2008). «NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type». Neurology 70 (1): 17-24. PMID 18003940. doi:10.1212/01.wnl.0000284596.95156.48.
- ↑ Craig MC, Murphy DG (2007). «Estrogen: effects on normal brain function and neuropsychiatric disorders». Climacteric. 10 Suppl 2: 97-104. PMID 17882683. doi:10.1080/13697130701598746.
- ↑ Mori K, Takeda M (2007). «Hormone replacement Up-to-date. Hormone replacement therapy and brain function». Clin Calcium (en japonés) 17 (9): 1349-1354. PMID 17767023.
- ↑ Birks J, Grimley Evans J (2007). «Ginkgo biloba for cognitive impairment and dementia». Cochrane Database Syst Rev (2): CD003120. PMID 17443523. doi:10.1002/14651858.CD003120.pub2. Consultado el 22 de febrero de 2008.
- ↑ «Alzhéimer: ¿qué juegos ayudan a prevenir el alzhéimer? Entrevista al Dr. Luis Ignacio Brusco.». 2019.
- ↑ Verghese J, Lipton R, Katz M, Hall C, Derby C, Kuslansky G, Ambrose A, Sliwinski M, Buschke H (2003). «Leisure activities and the risk of dementia in the elderly». N Engl J Med 348 (25): 2508-2516. PMID 12815136. doi:10.1056/NEJMoa022252.
- ↑ Bennett DA, Schneider JA, Tang Y, Arnold SE, Wilson RS (2006). «The effect of social networks on the relation between Alzheimer's disease pathology and level of cognitive function in old people: a longitudinal cohort study». Lancet Neurol 5 (5): 406-412. PMID 16632311. doi:10.1016/S1474-4422(06)70417-3.
- ↑ Bialystok E, Craik FIM, Freedman M (2007). «Bilingualism as a protection against the onset of symptoms of dementia». Neuropsychologia 42 (2): 459-464. doi:10.1016/j.neuropsychologia.2006.10.009.
- ↑ Davanipour Z, Tseng CC, Lee PJ, Sobel E (2007). «A case-control study of occupational magnetic field exposure and Alzheimer's disease: results from the California Alzheimer's Disease Diagnosis and Treatment Centers». BMC Neurol 7: 13. PMID 17559686. doi:10.1186/1471-2377-7-13.
- ↑ Qiu C, Fratiglioni L, Karp A, Winblad B, Bellander T (2004). «Occupational exposure to electromagnetic fields and risk of Alzheimer's disease». Epidemiology 15 (6): 687-694. PMID 15475717. doi:10.1097/01.ede.0000142147.49297.9d.
- ↑ Shcherbatykh I, Carpenter DO (mayo de 2007). «The role of metals in the etiology of Alzheimer's disease». J. Alzheimers Dis. 11 (2): 191-205. PMID 17522444.
- ↑ Rondeau V, Commenges D, Jacqmin-Gadda H, Dartigues JF (julio de 2000). «Relation between aluminum concentrations in drinking water and Alzheimer's disease: an 8-year follow-up study». Am. J. Epidemiol. 152 (1): 59-66. PMC 2215380. PMID 10901330.
- ↑ Kukull WA, Larson EB, Bowen JD, et al (junio de 1995). «Solvent exposure as a risk factor for Alzheimer's disease: a case-control study». Am. J. Epidemiol. 141 (11): 1059-1071; discusión: 1072-1079. PMID 7771442.
- ↑ Santibáñez M, Bolumar F, García AM (2007). «Occupational risk factors in Alzheimer's disease: a review assessing the quality of published epidemiological studies». Occupational and Environmental Medicine 64 (11): 723-732. PMID 17525096. doi:10.1136/oem.2006.028209.
- ↑ Seidler A, Geller P, Nienhaus A, Bernhardt T, Ruppe I, Eggert S, Hietanen M, Kauppinen T, Frölich L (2007). «Occupational exposure to low frequency magnetic fields and dementia: a case-control study». Occup Environ Med 64 (2): 108-114. PMID 17043077. doi:10.1136/oem.2005.024190.
- ↑ Rondeau V (2002). «A review of epidemiologic studies on aluminum and silica in relation to Alzheimer's disease and associated disorders». Rev Environ Health 17 (2): 107-121. PMID 12222737.
- ↑ Martyn CN, Coggon DN, Inskip H, Lacey RF, Young WF (mayo de 1997). «Aluminum concentrations in drinking water and risk of Alzheimer's disease». Epidemiology 8 (3): 281-286. PMID 9115023.
- ↑ Graves AB, Rosner D, Echeverria D, Mortimer JA, Larson EB (septiembre de 1998). «Occupational exposures to solvents and aluminium and estimated risk of Alzheimer's disease». Occup Environ Med 55 (9): 627-633. PMC 1757634. PMID 9861186.
- ↑ «UBA Salud». www.uba.ar. Archivado desde el original el 5 de junio de 2020. Consultado el 15 de noviembre de 2019.
- ↑ «Buscan desarrollar un test para predecir el alzhéimer». Fundación Instituto Leloir. 22 de septiembre de 2016. Consultado el 15 de noviembre de 2019.
- Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A. Sodium Butyrate Improves Memory Function in an Alzheimer's Disease Mouse Model When Administered at an Advanced Stage of Disease Progression. J Alzheimers Dis. (2011)
- Design of a comprehensive Alzheimer's disease clinic and research center in Spain to meet critical patient and family needs. M. Boada et. al.
Enlaces externos
editarWikimedia Commons alberga una categoría multimedia sobre Enfermedad de Alzheimer.
- Evolución en la investigación de la enfermedad de alzheimer
Datos: Q11081
Noticias: Categoría:Alzheimer