User:Smokefoot/sandbox
10.1016/0020-1693(94)04222-H V(II)en3 sulfate, Men3: one V, no Mo
M(en)3]OAc2 doi 10.1107/S2053229617006738 2.2693 2.2085 2.1661 2.1205 2.1693 2.1847 Mn-Zn Cu 4 x 2.05 2 x 2.39 Λ(δδδ) and Δ(λλλ)
10.1351/pac198456040491 Formation of metal complexes with ethylenediamine: a critical survey of equilibrium constants, enthalpy and entropy values
10.1039/c6cs00604c JAG
Perchlorate complexes
[edit]CuOAc 10.1039/C39730000124
{{[[Template:NMR spectroscopy |NMR spectroscopy ]]}}
AgClO4 (H2O): 10.1524/zkri.1995.210.11.879
To check in the future
[edit]mostly questionable homework:
{{Chembox | ImageFile = | ImageSize = | ImageAlt = | IUPACName = | OtherNames = |Section1={{Chembox Identifiers | CASNo = | CASNo_Comment = | CASNo1 = | CASNo1_Comment = | PubChem = | SMILES = }} |Section2={{Chembox Properties | Mo = 1|Cl = 3| O = 1 | MolarMass = | Appearance = | Density = 3.151 g/cm<sup>3</sup> | MeltingPt = | MeltingPt_notes = | BoilingPt = | BoilingPt_notes = | Solubility = }} |Section3={{Chembox Hazards | MainHazards = | FlashPt = | AutoignitionPt = }} }} Molybdenum oxytrichloride is the is the [[inorganic compound]] with the formula MoOCl<sub>3</sub>. It is a red paramagnetic solid, one of several [[oxychloride]]s of molybdenum. The compound is prepared by reduction of [[molybdenum oxotetrachloride]] with [[aluminium]]. One distinction for MoOCl<sub>3</sub> is that it crystallizes with terminal oxo and with bridging oxo ligands (as in NbOCl<sub>3</sub>).<ref>{{cite journal |doi=10.1039/J19690002019}}</ref> ==Structure== The compound has a polymeric structure consisting of Mo(O)Cl groups linked by chlorides. Thus, each Mo center is octahedral, but distorted. Two of the bridging chlorides have Mo-Cl distances near 2.45 Åbut the other two, which are trans to oxo ligands, are long and short, 2.78 and 2.34 Å. The one terminal chloride is bonded to Mo with a distance of 2.28 Å, the shortest bond.<ref>{{cite journal |doi=10.1039/j19700000022 |title=Crystal and Molecular Structure of Molybdenum(V) Oxide Trichloride |date=1970 |last1=Drew |first1=Michael G. B. |last2=Tomkins |first2=I. B. |journal=Journal of the Chemical Society A: Inorganic, Physical, Theoretical |page=22 }}</ref><ref>{{cite journal |doi=10.1039/j19690002415 |title=Oxide Chlorides of Tungsten and Molybdenum. Part II. Crystal Structure of the Monoclinic Form of Molybdenum Oxide Trichloride |date=1969 |last1=Ferguson |first1=G. |last2=Mercer |first2=M. |last3=Sharp |first3=D. W. A. |journal=Journal of the Chemical Society A: Inorganic, Physical, Theoretical |page=2415 }}</ref> ==Related compounds== [[Molybdenum dichloride dioxide]], a yellow diamagnetic solid. ==References== {{reflist}} {{Molybdenum compounds}} [[Category:Molybdenum(VI) compounds]] [[Category:Oxychlorides]]
Ref spam project
[edit]Zwitterion
[edit]- Novikov, Anton P.; Safonov, Alexey V.; German, Konstantin E.; Grigoriev, Mikhail S. (2023-12-01). "What kind of interactions we may get moving from zwitter to "dritter" ions: C–O⋯Re(O4) and Re–O⋯Re(O4) anion⋯anion interactions make structural difference between L-histidinium perrhenate and pertechnetate". CrystEngComm. doi:10.1039/D3CE01164J. ISSN 1466-8033.
Chlorine
[edit]- Wheeler, Ralph A.; Hoffmann, Roald. (1986-10). "A new magic cluster electron count and metal-metal multiple bonding". Journal of the American Chemical Society. 108 (21): 6605–6610. doi:10.1021/ja00281a025. ISSN 0002-7863.
{{cite journal}}: Check date values in:|date=(help) - [1]
- Zhang, Weiwei; Oganov, Artem R.; Goncharov, Alexander F.; Zhu, Qiang; Boulfelfel, Salah Eddine; Lyakhov, Andriy O.; Stavrou, Elissaios; Somayazulu, Maddury; Prakapenka, Vitali B.; Konôpková, Zuzana (2013-12-20). "Unexpected Stable Stoichiometries of Sodium Chlorides". Science. 342 (6165): 1502–1505. doi:10.1126/science.1244989. ISSN 0036-8075.
Sodium pertechnetate
[edit]- German, K. E.; Grigoriev, M. S.; Garashchenko, B. L.; Kopytin, A. V.; Tyupina, E. A. (2017-07-01). "Redetermination of the crystal structure of NaTcO4 at 100 and 296 K based on single-crystal X-ray data". Acta Crystallographica Section E: Crystallographic Communications. 73 (7): 1037–1040. doi:10.1107/S2056989017008362. ISSN 2056-9890. PMC 5499285. PMID 28775877.
{{cite journal}}: CS1 maint: PMC format (link)
Technetium
[edit]- Zhou, Di; Semenok, Dmitrii V.; Volkov, Mikhail A.; Troyan, Ivan A.; Seregin, Alexey Yu.; Chepkasov, Ilya V.; Sannikov, Denis A.; Lagoudakis, Pavlos G.; Oganov, Artem R.; German, Konstantin E. (2023-02-06). "Synthesis of technetium hydride $\mathrm{Tc}{\mathrm{H}}_{1.3}$ at 27 GPa". Physical Review B. 107 (6): 064102. doi:10.1103/PhysRevB.107.064102.
- German, K. E.; Peretrukhin, V. F.; Gedgovd, K. N.; Grigoriev, M. S.; Tarasov, A. V.; Plekhanov, Yu V.; Maslennikov, A. G.; Bulatov, G. S.; Tarasov, V. P.; Lecomte, M. (2005). "Tc Carbide and New Orthorhombic Tc Metal Phase". Journal of Nuclear and Radiochemical Sciences. 6 (3): 211–214. doi:10.14494/jnrs2000.6.3_211.
- Rimshaw, S. J. (1968). Hampel, C. A. (ed.). The Encyclopedia of the Chemical Elements. New York: Reinhold Book Corporation. pp. 689–693.
- Autler, S. H. (1968). "Technetium as a Material for AC Superconductivity Applications" (PDF). Proceedings of the 1968 Summer Study on Superconducting Devices and Accelerators. Retrieved 2009-05-05.
Group VII element
[edit]- German, Konstantin E.; Fedoseev, Alexander M.; Grigoriev, Mikhail S.; Kirakosyan, Gayane A.; Dumas, Thomas; Den Auwer, Christophe; Moisy, Philippe; Lawler, Keith V.; Forster, Paul M.; Poineau, Frederic (2021-09-24). "A 70‐Year‐Old Mystery in Technetium Chemistry Explained by the New Technetium Polyoxometalate [H 7 O 3 ] 4 [Tc 20 O 68 ] ⋅ 4H 2 O". Chemistry – A European Journal. 27 (54): 13624–13631. doi:10.1002/chem.202102035. ISSN 0947-6539.
Polyoxometalate
[edit]- Volkov, Mikhail A.; Novikov, Anton P.; Borisova, Nataliya E.; Grigoriev, Mikhail S.; German, Konstantin E. (10 August 2023). "Intramolecular Re···O Nonvalent Interactions as a Stabilizer of the Polyoxorhenate(VII)". Inorganic Chemistry. XX (XX): XXXX–XXXX. doi:10.1021/acs.inorgchem.3c01863. PMID XXX. S2CID XXXX.
{{cite journal}}: Check|pmid=value (help); Check|s2cid=value (help)
Rhenium
[edit]- Volkov, Mikhail A.; Novikov, Anton P.; Borisova, Nataliya E.; Grigoriev, Mikhail S.; German, Konstantin E. (2023-08-21). "Intramolecular Re···O Nonvalent Interactions as a Stabilizer of the Polyoxorhenate(VII)". Inorganic Chemistry. 62 (33): 13485–13494. doi:10.1021/acs.inorgchem.3c01863. ISSN 0020-1669.
Volkov, Mikhail A.; Novikov, Anton P.; Borisova, Nataliya E.; Grigoriev, Mikhail S.; German, Konstantin E. (2023-08-21). "Intramolecular Re···O Nonvalent Interactions as a Stabilizer of the Polyoxorhenate(VII)". Inorganic Chemistry. 62 (33): 13485–13494. doi:10.1021/acs.inorgchem.3c01863. ISSN 0020-1669.
Photo-nano-H2-
[edit]
MisterChemistry
[edit][1].
- User contributions for Dhansora210 spammer
- User:MisterChemistry
- User contributions for CuriousKemist ?
Phth
[edit]The question of how these plastics are affecting the population has arisen as plasticizer use in everyday items has increased. Additionally, if there are any disparities in how these plasticizers may affect minority populations and if they are more susceptible to complications. It has been found that exposure to phthalates is more likely in women and people of color.[14] In one study, researchers looked into potential differences between gender and race as well as potential consequences of this higher phthalate exposure. The study paid particular attention to the relationship between urinary phthalate metabolites and risk factors for diabetes in individuals with no previous diabetes diagnosis. It was found that while there were no statistically significant differences between men and women overall, there were differences between Mexican-Americans, blacks, and whites in terms of the overall risk of disturbance of glucose homeostasis. With Mexican-Americans having a fasting blood glucose (FBG) increase of 5.82 mg/dL, blacks having a fasting blood glucose increase of 3.63 mg/dL, and whites having a fasting blood glucose increase of 1.79 mg/dL, there was evidence of an increased risk for minorities.[14] Overall, the study concludes that phthalates may alter glucose homeostasis and insulin sensitivity, and that different populations may be more severely impacted. Higher levels of some phthalate metabolites were associated with elevated FBG, fasting insulin, and insulin resistance. In a different study looking at the presence of phthalate metabolites in pregnant women, it was found that non-Hispanic black women and Hispanic women had higher levels of some phthalate metabolites, indicating potential racial disparities in pregnancy outcomes.[15]
Population info W hemis
[edit]| Country | Total Fertility rate in 2021 (births/woman) |
population (M) | area mi2 | Pop density | per cap income (k, PPP) |
|---|---|---|---|---|---|
| Haiti | 2.81 | 8.8 | 10 | 989 | 3.2 |
| Bolivia | 2.62 | 12.1 | 424 | 27 | 10.3 |
| Paraguay | 2.47 | 6.1 | 157 | 39 | 15.5 |
| Guyana | 2.40 | 0.79 | 83 | 9.1 | 61 |
| Guatemala | 2.40 | 18 | 42 | 334 | 10.6 |
| Honduras | 2.36 | 9.6 | 43 | 220 | 7.2 |
| Suriname | 2.35 | 0.632 | 63 | 18.3 | |
| Panama | 2.33 | 4.3 | 29 | 145 | 42.7 |
| Nicaragua | 2.32 | 6.4 | 50 | 132 | 7.6 |
| Dominican Republic | 2.27 | 11.4 | 18.7 | 570 | 25.5 |
| World | 2.27 | ||||
| Venezuela | 2.21 | 30.5 | 354 | 87 | 8 |
| Peru | 2.19 | 33.5 | 1285 | 60 | 15.9 |
| Population replacement | 2.10 | ||||
| Ecuador | 2.03 | 17.4 | 109 | 179 | 13.3 |
| Belize | 2.01 | 0.44 | 8.9 | 46 | 11.1 |
| Argentina | 1.89 | 47.3 | 1073 | 37 | 26.5 |
| Mexico | 1.82 | 130 | 761 | 158 | 25 |
| El Salvador | 1.80 | 6.6 | 8.1 | 840 | 11.7 |
| Colombia | 1.72 | 52.156 | 440 | 118 | 19.5 |
| United States | 1.66 | 379.6 | 335 | 34 | 80 |
| Brazil | 1.64 | 203 | 3.287 | 65 | 20 |
| Chile | 1.54 | 18,549 | 292 | 62 | 29.9 |
| Costa Rica]] | 1.53 | 5 | 19.7 | 85 | 27 |
| Uruguay | 1.49 | 3.44 | 68 | 50.5 | 28.9 |
| Russia | 1.49 | 147 | 6.6 | 22 | 35.3 |
| Cuba | 1.44 | 11 | 48.2 | 264 | ?? |
| Jamaica | 1.35 | 2734 | 4.2 | 689 | 13 |
|}==Other== 4+3] CYCLOADDITION IN WATER. SYNTHESIS OF 2,4-endo,endo-DIMETHYL-8-OXABICYCLO[3.2.1]OCT-6-EN-3-ONE Mark Lautens and Giliane Bouchain Org. Synth. 2002, 79, 251 DOI: [3] [4 + 3] CYCLOADDITION OF AMINOALLYL CATIONS WITH 1,3-DIENES: 11-OXATRICYCLO[4.3.1.12,5]UNDEC-3-EN-10-ONE Jonghoon Oh, Chewki Ziani-Cherif, Jong-Ryoo Choi, and Jin Kun Cha, Org. Synth. 2002, 78, 212 DOI:
Marginal cruft
[edit]- Lithium beryllide, fiction supported by theory.
- [[Urea-formaldehyde}} belabored description of emissions.
MG chem thing
[edit]- Cotton and Wilkinson's Advanced Inorganic Chemistry" (6th Edition): "Part 1 Survey of Principles", "Part 2 The Chemistry of the Main Group Elements", etc
- Shriver and Atkins Inorganic Chemistry (5th Edition): MG chem not in index. "Nonmetal" refers to p 262 showing the periodic table highlighting elements above the B-Po diagonal.
- Elschenbroich's "Organometallic Chemistry" main sections: "Introduction" (chapters 1-3), "Main-Group Organometallics" (chapters 4-11), "Organometallic Chemistry of the Transition Metals" (chapters 12-18)
Notes and projects
[edit]Hexamethylenetetramine Alpha hydroxy acid, ck sources, list main examples
Quinolizine is a heterocyclic organic compound with the formula (C4H4)2CHNH. A colorless solid, it is one of the isomers of dihydroquinoline, but with a secondary amine at the bridge site. The saturated derivative of quinolizine is quinolizidine.
A transition metal diamine complex is a coordination compound containing one or more diamine ligands. Such complexes played a role in the development of coordination chemistry, particularly with regards to the stereochemistry of chelate rings.
Diamine ligands
[edit]The principal diamine ligand is ethylenediamine (1,2-diaminoethane, abbreviated en). It is the premier and simplest diamine ligand, which enjoys utility outside of coordination chemistry. Related diamines include:
- propylenediamine (1,2-diaminopropane, pn), which is notable as being chiral.
- trimethylenediamine (1,3-diaminopropane, tn), which forms 6-membered MN2C3 chelate rings.
- trans-1,2-diaminocyclohexane (chxn), which is notable as being chiral but also C2-symmetric.
- tetramethylethylenediamine (tmeda), a di-tertiaryamiine that is widely used to form complexes alkali and alkaline earth metal.
Homoleptic complexes
[edit]Derivatives of en, pn, tn, chxn
[edit]Numerous homoleptic complexes are known of the type [M(diamine)3]n+ M = Cr(III), Co(III), Rh(III), Rh(III), Ir(III), Ni(II), amd Pt(IV). One specific example is found in the hydrated salt [Co(chxn)3]Cl3.[4] Pd(II), Pt(II), and Au(III) form homoleptic complexes of the type [M(diamin)2]n+.
References
[edit]- ^ Knepper, T. (2003). "Synthetic chelating agents and compounds exhibiting complexing properties in the aquatic environment". Trac Trends in Analytical Chemistry. 22 (10): 708–724. doi:10.1016/S0165-9936(03)01008-2.
- ^ Cite error: The named reference
autowas invoked but never defined (see the help page). - ^ "[4 + 3] Cycloaddition of Aminoallyl Cations with 1,3-Dienes: 11-Oxatricyclo[4.3.1.12,5]Undec-3-En-10-One". Organic Syntheses. 78: 212. 2002. doi:10.15227/orgsyn.078.0212.
- ^ Marumo, F.; Utsumi, Y.; Saito, Y. (1970). "The crystal structure of (−)589-tris[(+)-trans-1,2-diaminocyclohexane]cobalt(III) chloride pentahydrate, (−)589-{Co[(+)-CHXN]3}Cl3.5H2O". Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry. 26 (10): 1492–1498. doi:10.1107/S0567740870004387.
Über die Einwirkung von Quecksilbersalzen auf Eisenpentacarbonyl
H. Hock, H. Stuhlmann First published: 10. Oktober 1928 https://doi.org/10.1002/cber.19280610907 Volume61, Issue9 10. Oktober 1928 Pages 2097-2101
In chemistry, persulfide refers to the functional group R-S-S-H.[1] Persulfides are intermediates in the biosynthesis of iron-sulfur proteins and are invoked a precursors to hydrogen sulfide, a signalling molecule.
Compared to thiols (R-S-H), persulfides are uncommon. They are thermodynamically unstable with respect to loss of elemental sulfur:
- RSSH RSH + 1/8S8
Nonetheless they are kinetically stable.
The S-H bond is both more acidic and more fragile than in thiols. The bond dissociation energy is 22 kcal/mol weaker than a thiol. This effect is attributed to the stability of the RSS. radical. The pKa is 6.2 is much lower than the pKa of 7.5 for thiols. Thus persulfides exist predominantly in the ionized form at neutral pH.
References
[edit]Mann, B. E.; Taylor, B. E. I3C NMR Data for Organometallic Compounds·, Academic Press: London, 1981 isbn 0124691501
ZrBrx
[edit]ZrBr3 density 4.45 10.1021/ic50154a056 doi
ZrBr Corbett 0.1021/ic50174a041
References
[edit]Chemical compoundss
[edit]
Manganese is renown for adopting many oxidation states, ranging from −3 to +7.
Halides
[edit]The dihalides of Mn are common reagents.
Oxides
[edit]Manganese dioxide is the most important compound as it comprises the main mineral and a component of some batteries. Alkali metal salts of manganate (MnO42-) and permanganate (MnO4-) are deep green and violet. Other more esoteric oxidees are the oxyhalides (MnO3F and MnO3Cl) and the explosive molecule Mn2O7.
CAS Registry Number 7450-69-3 ~293~46 C6 H9 N2 O2 P Phosphorodiamidic acid, phenyl ester Molecular Weight 172.12 Melting Point (Experimental) Value: 185 °C
CdTe MoCl3 and bromide and iodide Molybdenum(II) bromide
| Names | |
|---|---|
| Other names
Benzo[g]quinoline
| |
| Identifiers | |
| Properties | |
| Density | 1.329 g/cm3[3] |
| Melting point | 247 °C (477 °F; 520 K) |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
| |
1-Azaanthracene is one of several isomers of benzoquinoline. A colorless solid, it is prepared by cyclization of 2-benzyl derivative of nicotinaldehyde.[4]
Violanthronequinone is prepared by coupling of benzanthrone to 4,4-bibenzanthronyl. The latter is oxidative ring closure to violanthrone, which sustains oxidation to give the violanthronequinone. Reduction with sodium hydrogen sulfite give 16,17-dihydroxyviolanthrone, O-methylation of which gives the dimethoxy diol called Caledon Jade Green.[5]==Ln2O3== M2O3 is a basic oxide. Insoluble in water, this ...-colored solid absorbs atmospheric water to form the trihydroxide. It dissolve in acidic water to form aquo complex [M(H2O)n]3+. The oxide is produced both by burning the metal or roasting the hydroxides or calcining the carbonate.
It adopts a ... structure according to X-ray crystallography. As such each M is seven-coordinate.
Inorganic esters
[edit]
Esters can also be derived from inorganic acids.
- Phosphoric acid forms phosphate esters, e.g. triphenylphosphate
- sulfuric acid forms sulfate esters, e.g., dimethylsulfate
- nitric acid forms nitrate esters, e.g. methyl nitrate
- boric acid forms borates, e.g. trimethylborate.
- carbonic acid forms carbonate esters, e.g. ethylene carbonate
Inorganic acids that exist as tautomers form diverse esters
- orthophosphorous acid forms two kinds of phosphite esterss, e.g. triethylphosphite (P(OEt)3) and diethylphosphite (HP(O)(OEt)2).
Inorganic acids that are unstable or elusive form stable esters.
- chromic acid, which has never been detected, forms di-tert-butyl chromate
- sulfurous acid, which is rare, forms dimethylsulfite.
TheFreeDictionary
[edit]to replace ref Propenyl Synthetic element Glycolic acid Hydrate Supermolecule Mercapturic acid Glucose-6-phosphate dehydrogenase deficiency Calcium hydride Isotopes of oxygen Germanide Prostacyclin A68 protein CS gas Hugo Schiff Amphotericin B Butyl nitrate Flow cytometry Chalcogen
Various
[edit]
"Mono Carbamate Protection of Aliphatic Diamines Using Alkyl Phenyl Carbonates". Organic Syntheses. 84: 209. 2007. doi:10.15227/orgsyn.084.0209. MONO CARBAMATE PROTECTION OF ALIPHATIC DIAMINES USING ALKYL PHENYL CARBONATES Michael Pittelkow, Rasmus Lewinsky, and Jørn B. Christensen Org. Synth. 2007, 84, 209 DOI:
?
[edit]rac -59 °C Boiling Point (Experimental) Value: 188.2 °C Density (Experimental) Value: 1.036 g/cm3 | Condition: Temp: 25 °C
4254-15-3 (2S)-1,2-Propanediol, Molecular Weight Boiling Point (Experimental) Value: 88.7 °C | Condition: Press: 13 Torr Density (Experimental) Value: 1.0364 g/cm3 | Condition: Temp: 20 °C
4254-14-2 = R enantiomer
SES
[edit]Joshua R. Sacher and Steven M. Weinreb "DISCUSSION ADDENDUM for: 2-Trimethylsilylethanesulfonyl Chloride (SES-Cl) Org. Synth. 2012, 89, 34-43. doi:10.15227/orgsyn.089.0034
OM chem
[edit]The roots of the term "organometallic", "organo-" and "metallic" are convenient starting points for defining the scope of this family of compounds. As mentioned above, the metal can include any metallic element. The organic ligands more diverse, but can be classified as follows:
- alkyl and aryl ligands.
Formally viewed as anions, alkyl and aryl ligands are the quintessential organic ligands and such compounds find pervasive applications. The parent alkyl and aryl ligands are methyl (CH3) and phenyl (C6H5). Homoleptic complexes, consisting only of such ligands, include hexamethyltungsten and triphenylaluminium. More commonly, alkyl and aryl ligands are found in mixed ligand complexes e.g., dicyclopentadienyltitanium dimethyl and PhMgBr(OEt2)2. Beyond methyl and phenyl, myriad other alkyl and aryl ligands are known<ref name= Elschenbroich/>
- alkene ligands.
Formally viewed as 2e donor ligands, alkenes form diverse complexes and are important substrates for many practical processes. The parent ligand is ethylene, C2H4. Incidentally ethylene is the feedstock for the production of polyethylene, which in turn is catalyzed by organometallic compounds. Homoleptic complexes of ethylene are known, such as tris(ethylene)nickel(0). More commonly, ethylene as a ligand is encountered in mixed ligand complexes, such as chlorobis(ethylene)rhodium dimer. Beyond ethylene, many alkenes exhibit affinity for metal centers. Also common are diene, triene, and even tetraene ligands. Examples include bis(cyclooctatetraene)nickel(0) and bis(η8-cyclooctatetraenyl)uranium(IV). Special subset of triene-like ligands are arenes, which most commonly bind metals via all six carbon centers.
- combinations of alkyl and alkene groups give rise to diverse families of anionic ligands including allyl (C3H5) and cyclopentadienyl (C5H5) ligands, as well as substituted derivatives. Examples include allylpalladium chloride dimer and ferrocene.
- alkyne ligands
Formally viewed as 2e or, more rarely, 4e, Lewis bases, alkynes readily form adducts with transition metals. The parent ligand is acetylene (C2H2). Homoleptic complexes, consisting only of such ligands, includes W(C2Ph2)3. More commonly, alkynes are found in mixed ligand complexes e.g., Pt(PPh3)2(C2Ph2)(CO)6 and the tetrahedrane Co2(C2H2)(CO)6 Beyond acetylene itself, myriad other derivatives are known, even including unstable alkynes such as dichloroacetylene<ref name= Elschenbroich/>
- alkylidene ligands
Formally viewed as 2e Lewis bases, a variety of alkylidenes form adducts with transition metals. The parent ligand is methylene (CH2), a highly reactive entity in the absence of its complexes Homoleptic complexes, consisting only of alkylidene, includes derivatives of N-heterocyclic carbenes, such as Pd(IMes)2. More commonly, carbenes are found in mixed ligand complexes e.g., W(CO)5)CPh2.
- alkylidyne ligands
- carbido ligands
Carbon atoms are not stable in the condensed phase, but many carbides are known usually as refractory solids. Many molecular complexes of "C" are known but in almost all cases, the carbon is a bridging ligand. Examples include Fe6C(CO)16]2-, Fe5C(CO)15, and Fe4C(CO)14. In rare cases, complexes are known featuring a terminal carbido ligand, e.g.
- saturated hydrocarbons
Saturated hydrocarbons are important ligands. They weakly basic and most such complexes have been fully characterized only when the interaction is part of a chelating ligand. Unlike other ligands in this list, agostic interactions are a form of 3-center, 2-electron bonding.
Molecular sieve
[edit]

A molecular sieve is a material with pores (very small holes) of uniform size. These pore diameters are of the dimensions of small molecules, thus large molecules cannot be absorbed, while smaller molecules can. Many molecular sieves are used as desiccants. Some examples include activated charcoal and silica gel.<ref>{{cite web|url=http://chemistry.about.com/od/chemistryglossary/g/Molecular-Sieve-Definition.htm |title=Molecular Sieve Definition - Definition of Molecular Sieve - What Is a Molecular Sieve? |publisher=Chemistry.about.com |date=2013-12-18 |accessdate=2014-02-26}}</ref>
Porosity
[edit]The diameter of a molecular sieve is measured in ångströms (Å) or nanometres (nm). According to IUPAC notation, microporous materials have pore diameters of less than 2 nm (20 Å) and macroporous materials have pore diameters of greater than 50 nm (500 Å); the mesoporous category thus lies in the middle with pore diameters between 2 and 50 nm (20–500 Å).<ref>{{cite journal|author=J. Rouquerol |display-authors=etal |title=Recommendations for the characterization of porous solids (Technical Report)|journal=Pure & Appl. Chem|volume=66|date=1994|pages=1739–1758|url=http://www.iupac.org/publications/pac/66/8/1739/pdf/|format=free download pdf|doi=10.1351/pac199466081739|issue=8}}</ref> Molecular sieves are microporous.
Materials
[edit]Zeolites
[edit]Zeolites are the most common molecular sieves. They consist of anionic aluminosilicate frameworks, where the charges are neutralized by protons and alkali metal and alkaline earth cations. Because of the uniformity and rigidity of their frameworks, zeolitic molecular sieves "discriminate, and organize molecules with precisions that can be less than 1 Å."<ref>Mark E. Davis, Raul F. Lobo "Zeolite and molecular sieve synthesis" Chem. Mater., 1992, 4 (4), pp 756–768.{{DOI|10.1021/cm00022a005}}</ref>
| zeolite type | pore size in angstroms | approximate formula | typical application |
|---|---|---|---|
| zeolite A | 3.8 | Na12[(AlO2)12(SiO2)12 | desiccant for natural gas |
| zeolite A | 4.4 | Ca5Na2[(AlO2)12(SiO2)12 | separation of alkanes |
| zeolite A | 2.9 | K12[(AlO2)12(SiO2)12 | desiccant for ethylene |
| zeolite X | 8.0 | Sr21Ba22[(AlO2)86(SiO2)106 | separation of xylene isomers |
Hydrolysis
[edit]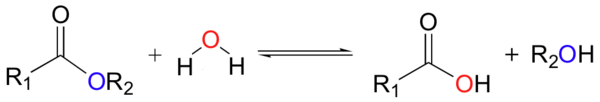
Hydrolysis (/haɪˈdrɒlɪsɪs/; from Greek hydro- 'water' and lysis 'to unbind') usually means the cleavage of chemical bonds by the addition of water.
Hydrolysis can be the reverse of a condensation reaction in which two molecules join together into a larger one and eject a water molecule. Thus hydrolysis adds water to break down, whereas condensation builds up by removing water. Often, hydrolysis or saccharification is a step in the degradation of a substance
| Identifiers | |
|---|---|
| Properties | |
| C3H3Cl3O2 | |
| Molar mass | 177.41 g·mol−1 |
| Appearance | colorless liquid |
| Density | 1.515 g/cm3 |
| Melting point | −17.5 °C (0.5 °F; 255.7 K) |
| Boiling point | 153.8 °C (308.8 °F; 426.9 K) |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
| |
Methyl trichloroacetate is the organic compound with the formula Cl3CCO2CH3.==Examples==
Esters and amides
[edit]Esters undergo hydrolysis to give alcohols and carboxylic acids:
These reactions can be accelerated by acids and bases. Enzymes called esterases catalyze this reaction.
Acid–base-catalysed hydrolyses are very common; one example is the hydrolysis of amides or esters. Their hydrolysis occurs when the nucleophile (a nucleus-seeking agent, e.g., water or hydroxyl ion) attacks the carbon of the carbonyl group of the ester or amide. In an aqueous base, hydroxyl ions are better nucleophiles than polar molecules such as water. In acids, the carbonyl group becomes protonated, and this leads to a much easier nucleophilic attack. The products for both hydrolyses are compounds with carboxylic acid groups.
Salts
[edit]A common kind of hydrolysis occurs when a salt of a weak acid or weak base (or both) is dissolved in water. Water spontaneously ionizes into hydroxide anions and hydronium cations. The salt also dissociates into its constituent anions and cations. For example, sodium acetate dissociates in water into sodium and acetate ions. Sodium ions react very little with the hydroxide ions whereas the acetate ions combine with hydronium ions to produce acetic acid. In this case the net result is a relative excess of hydroxide ions, yielding a basic solution.
Strong acids also undergo hydrolysis. For example, dissolving sulfuric acid (H2SO4) in water is accompanied by hydrolysis to give hydronium and bisulfate, the sulfuric acid's conjugate base. For a more technical discussion of what occurs during such a hydrolysis, see Brønsted–Lowry acid–base theory.
Perhaps the oldest commercially practiced example of ester hydrolysis is saponification (formation of soap). It is the hydrolysis of a triglyceride (fat) with an aqueous base such as sodium hydroxide (NaOH). During the process, glycerol is formed, and the fatty acids react with the base, converting them to salts. These salts are called soaps, commonly used in households.
However, proteases do not catalyse the hydrolysis of all kinds of proteins. Their action is stereo-selective: Only proteins with a certain tertiary structure are targeted as some kind of orienting force is needed to place the amide group in the proper position for catalysis. The necessary contacts between an enzyme and its substrates (proteins) are created because the enzyme folds in such a way as to form a crevice into which the substrate fits; the crevice also contains the catalytic groups. Therefore, proteins that do not fit into the crevice will not undergo hydrolysis. This specificity preserves the integrity of other proteins such as hormones, and therefore the biological system continues to function normally.
Upon hydrolysis, an amide converts into a carboxylic acid and an amine or ammonia (which in the presence of acid are immediately converted to ammonium salts). One of the two oxygen groups on the carboxylic acid are derived from a water molecule and the amine (or ammonia) gains the hydrogen ion. The hydrolysis of peptides gives amino acids.
Mechanism for acid-catalyzed hydrolysis of an amide.

Many polyamide polymers such as nylon 6,6 hydrolyse in the presence of strong acids. The process leads to depolymerization. For this reason nylon products fail by fracturing when exposed to small amounts of acidic water. Polyesters are also susceptible to similar polymer degradation reactions. The problem is known as environmental stress cracking.
ATP
[edit]ATP hydrolysis is highly exothermic. The energy released in this process supplies energy for many biological processes.
Hydrolysis is related to energy metabolism and storage. All living cells require a continual supply of energy for two main purposes: the biosynthesis of micro and macromolecules, and the active transport of ions and molecules across cell membranes. The energy derived from the oxidation of nutrients is not used directly but, by means of a complex and long sequence of reactions, it is channelled into a special energy-storage molecule, adenosine triphosphate (ATP). The ATP molecule contains pyrophosphate linkages (bonds formed when two phosphate units are combined together) that release energy when needed. ATP can undergo hydrolysis in two ways: the removal of terminal phosphate to form adenosine diphosphate (ADP) and inorganic phosphate, or the removal of a terminal diphosphate to yield adenosine monophosphate (AMP) and pyrophosphate. The latter usually undergoes further cleavage into its two constituent phosphates. This results in biosynthesis reactions, which usually occur in chains, that can be driven in the direction of synthesis when the phosphate bonds have undergone hydrolysis.
Polysaccharides
[edit]
Carbohydrates undergo hydrolysis. In the presence of water, they break down into its component sugars. For example, sucrose hydrolyzes into glucose and fructose). This process is termed saccharification. Enzymes that hydrolyse glycosidic bonds are called "glycoside hydrolases" or "glycosidases". Lactase is essential for digestive hydrolysis of lactose in milk; many adult humans do not produce lactase and cannot digest the lactose in milk (not a disorder).
Malt made from barley is used as a source of β-amylase to break down starch into the disaccharide maltose, which can be used by yeast to produce beer. Other amylase enzymes may convert starch to glucose or to oligosaccharides. Cellulose is first hydrolyzed to cellobiose by cellulase and then cellobiose is further hydrolyzed to glucose by beta-glucosidase. Animals such as cows (ruminants) are able to hydrolyze cellulose into cellobiose and then glucose because of symbiotic bacteria that produce cellulases.
Metal aqua ions
[edit]Metal ions are Lewis acids, and in aqueous solution they form metal aqua ions of the general formula M(H2O)nm+.<ref>{{Cite book |author=Burgess, J. |year=1978 |title=Metal ions in solution |location=New York |publisher=Ellis Horwood}}</ref><ref>{{cite book | last = Richens | first =D. T. | title = The chemistry of aqua ions: synthesis, structure, and reactivity: a tour through the periodic table of the elements | publisher = Wiley | year = 1997 | isbn = 0-471-97058-1}}</ref> The aqua ions undergo hydrolysis, to a greater or lesser extent. The first hydrolysis step is given generically as
- M(H2O)nm+ + H2O ⇌ M(H2O)n−1(OH)(m−1)+ + H3O+
Thus the aqua cations behave as acids in terms of Brønsted-Lowry acid-base theory. This effect is easily explained by considering the inductive effect of the positively charged metal ion, which weakens the O-H bond of an attached water molecule, making the liberation of a proton relatively easy.
The dissociation constant, pKa, for this reaction is more or less linearly related to the charge-to-size ratio of the metal ion.<ref name="bm">Baes, C.F.; Mesmer, R.E. ''The Hydrolysis of Cations'', (1976), Wiley, New York</ref> Ions with low charges, such as Na+ are very weak acids with almost imperceptible hydrolysis. Large divalent ions such as Ca2+, Zn2+, Sn2+ and Pb2+ have a pKa of 6 or more and would not normally be classed as acids, but small divalent ions such as Be2+ undergo extensive hydrolysis. Trivalent ions like Al3+ and Fe3+ are weak acids whose pKa is comparable to that of acetic acid. Solutions of salts such as BeCl2 or Al(NO3)3 in water are noticeably acidic; the hydrolysis can be suppressed by adding an acid such as nitric acid, making the solution more acidic.
Hydrolysis may proceed beyond the first step, often with the formation of polynuclear species via the process of olation.[6] Some "exotic" species such as Sn3(OH)42+<ref>{{greenwood&Earnshaw|page=384}}</ref> are well characterized. Hydrolysis tends to proceed as pH rises leading, in many cases, to the precipitation of a hydroxide such as Al(OH)3 or AlO(OH). These substances, major constituents of bauxite, are known as laterites and are formed by leaching from rocks of most of the ions other than aluminium and iron and subsequent hydrolysis of the remaining aluminium and iron.
reminders
[edit]https://en.wikipedia.org/wiki/Special:Contributions/Teun71 https://en.wikipedia.org/wiki/Special:Contributions/Isabelabicalho Polyhydroxybutyrate Methane CAS 109-01-3 NMe piperazine 15400 refs Piperazine, 1-methyl- mp -6 °C bp 138 °C Density 0.89928 g/cm3 505-66-8 1248 refs
5687-07-0 DACO 78-80 °C only 76 refs
6572-95-8 DTO bp 43-46 °C 10 Torr 4% of S(CH3CH2CH2)2S, b. 245-6°, m. -15° Meadow, J. R.; Reid, E. E., "Ring compounds and polymers from polymethylene dihalides and dimercaptans", J. Am. Chem. Soc. 1934, 56, 2177-2180. 10.1021/ja01325a058
- ^ Park, Chung-Min; Weerasinghe, Laksiri; Day, Jacob J.; Fukuto, Jon M.; Xian, Ming (2015). "Persulfides: Current knowledge and challenges in chemistry and chemical biology". Molecular Biosystems. 11 (7): 1775–1785. doi:10.1039/c5mb00216h.
- ^ "Ch. 20". Shriver and Atkins' Inorganic Chemistry. Oxford University Press. 2010. ISBN 978-0-19-923617-6.
- ^ Yamada, Shinji; Kawamura, Chiaki (2012). "[4 + 4] Photodimerization of Azaanthracenes in Both Solution and Solid Phase Controlled by Cation−π Interactions". Organic Letters. 14 (6): 1572–1575. doi:10.1021/ol3003089.
- ^ Krapcho, A. Paul; Gilmor, Timothy P. (1999). "General preparative route to benzo[g]quinolines (1-azaanthracenes)". Journal of Heterocyclic Chemistry. 36 (2): 445–452. doi:10.1002/jhet.5570360218.
- ^ Bien, H.-S.; Stawitz, J.; Wunderlich, K. (2005). "Anthraquinone Dyes and Intermediates". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a02_355. ISBN 978-3527306732.
- ^ Cite error: The named reference
bmwas invoked but never defined (see the help page).