Usherov sindrom
| Usherov sindrom (Usher–Hallgrenov sindrom) | |
|---|---|
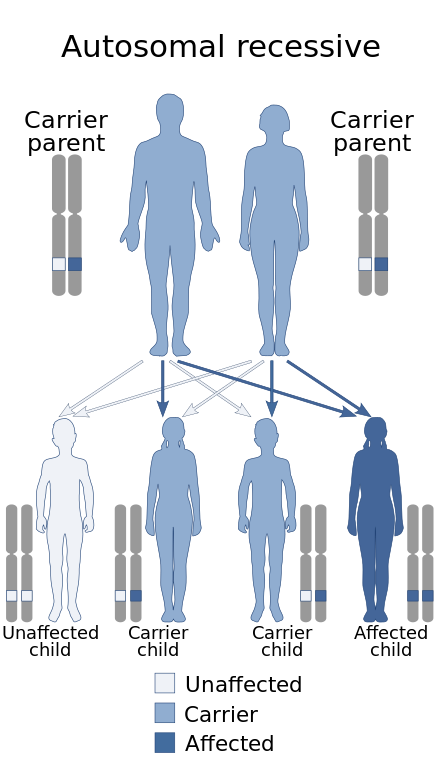 Usherov sindrom nasljeđuje se kao autosomno recesivno obilježje | |
| Klasifikacija i vanjski resursi | |
| ICD-10 | H35.53 |
| OMIM | 276900 OMIM: 276901 |
| DiseasesDB | 13611 |
| MeSH | D052245 |
Usherov sindrom, znan i kao Hallgrenov sindrom, Usher–Hallgrenov sindrom, retinitis pigmentosa–dysacusis syndrome ili dystrophia retinae dysacusis syndrome,[1] je rijetki genetički poremećaj uzrokovan mutacijom bilo kojeg od najmanje 11 gena, što rezultira kombinacijom gluhoće i sljepila. To je glavni uzrok gluhoslijepoća i do sada je neizlječiv.
Usherov sindrom klasificiran je u tri podtipa (I, II i III), prema odgovornim genima i nastupu gluhoće. Sva tri podtipa uzrokovana su mutacijama gena koji su uključeni u funkciju unutrašnjeg uha i mrežnjače. Ove mutacije se nasljeđuju u autosomno recesivnom obrascu.
Tipovi
[uredi | uredi izvor]Usherov sindrom I
[uredi | uredi izvor]Ljudi s poremećajem I rođeni su duboko gluhi i u prvoj deceniji života počinju gubiti vid. Oni takođe imaju poteškoće u ravnoteži i uče da polahko hodaju kao djeca, zbog problema u vestibulskom sistemu.
Usherov sindrom tipa I može biti uzrokovan mutacijama bilo kojeg od nekoliko različitih gena: CDH23, MYO7A, PCDH15, USH1C i USH1G . Ovi geni funkcioniraju u razvoju i održavanju struktura unutrašnjeg uha, kao što su trepljaste ćelije (stereocilie), koje prenose zvuk i signale pokreta u mozak. Promjene u ovim genima mogu uzrokovati nemogućnost održavanja ravnoteže (vestibulska disfunkcija) i gubitak sluha. Geni također imaju ulogu u razvoju i stabilnosti mrežnjače, utičući na strukturu i funkciju i štapičastih fotoreceptorskih i potpornih ćelija zvanih mrežnjačni pigmentirani epitel. Mutacije koje utiču na normalnu funkciju ovih gena mogu rezultirati bolešćuj retinitis pigmentosa i rezultirajućim gubitkom vida.
Procijenjena prevalencija Usherovog sindroma tipa I, u opštoj populaciji širom svijeta, je 3 do 6 na 100.000 ljudi. Utvrđeno je da je tip I češći kod ljudi porijeklom aškenazijskih Jevreja (srednje i istočne Evrope) i kod francuskog stanovništva – akadijske populacije (Louisiana, SAD).
Usherov sindrom II
[uredi | uredi izvor]Ljudi s Usherovim sindromom II nisu rođeni gluhi i uglavnom su nagluhi, a ne gluhi, a sluh im se vremenom ne pogoršava; šta više, čini se da nemaju primjetnih problema s ravnotežom. Oni također počinju gubiti vid kasnije (u drugoj deceniji života) i mogu sačuvati vid čak i u srednjim godinama.
Usherov sindrom tipa II može biti uzrokovan mutacijama bilo kojeg od tri različita gena: USH2A, GPR98 i DFNB31 . Protein kodiran genom USH2A, usherin, nalazi se u potpornom tkivu u unutrašnjem uhu i mrežnjači. Usherin je presudan za pravilan razvoj i održavanje ovih struktura, što može pomoći u objašnjavanju njegove uloge u gubitku sluha i vida. Položaj i funkcija druga dva proteina još nisu poznati.
Usherov sindrom tip II javlja se najmanje onoliko često koliko i tip I, ali budući da tip II može biti nedijagnosticiran ili ga je teže otkriti, mogao bi biti i do tri puta češći od tipa I.
Usherov sindrom III
[uredi | uredi izvor]Ljudi s Usherovim sindromom III ne rađaju se gluhi, već imaju progresivni gubitak sluha, a otprilike polovina ima poteškoće u ravnoteži.
Mutacije samo jednog gena, CLRN1 , povezane su s Usherovim sindromom tipa III. On kodira klarin-1, protein važan za razvoj i održavanje unutrašnjeg uha i mrežnjače. Međutim, funkcija proteina u tim strukturama i kako njegova mutacija uzrokuje gubitak sluha i vida još uvijek je slabo razumljiva.
Učestalost Usherovog sindroma tipa III značajna je samo u [[Finci|finskoj populaciji[2] i osoba jevrejskog porijrkla Aškenazija. Rijetko je primijećen u nekoliko drugih etničkih grupa.
Simptomi
[uredi | uredi izvor]Usherov sindrom karakterizira gubitak sluha i postupno oštećenje vida. Gubitak sluha uzrokovan je oštećenjem unutrašnjeg uha, dok je gubitak vida posljedica retinitis pigmentosa (RP), degeneracije ćelija mrežnjače. Obično se prvo pogađaju štapiće u mrežnjačama, što dovodi do ranog noćnog sljepila (niktalopija) i postepenog gubitka perifernog vida. U drugim slučajevima dolazi do rane degeneracije čepića u makuli, što dovodi do gubitka centralne oštrina. U nekim slučajevima, fovea i vid je pošteđen, što dovodi do "vizije krafne"; centralni i periferni vid su netaknuti, ali anulus postoji oko centralne regije u kojoj je vid oštećen.
Uzrok
[uredi | uredi izvor]| Tip | Freq[3] | Genski lokus | Gen | Protein | Funkcija | Veličina (AA) | UniProt | OMIM |
|---|---|---|---|---|---|---|---|---|
| USH1B | 39–55% | 11q13.5 | MYO7A | Miozin VIIA | Motorni protein | 2215 | Q13402 | OMIM: 276900 |
| USH1C | 6–7% | 11p15.1-p14 | USH1C | Harmonin | PDZ-domenski protein | 552 | Q9Y6N9 | OMIM: 276904 |
| USH1D | 19–35% | 10q21-q22 | CDH23 | Kadherin 23 | Adhezija ćelija | 3354 | Q9H251 | OMIM: 601067 |
| USH1E | Rijedak | 21q21 | ? | ? | ? | ? | ? | OMIM: 602097 |
| USH1F | 11–19% | 10q11.2-q21 | PCDH15 | Protokadherin 15 | Adhezija ćelija | 1955 | Q96QU1 | OMIM: 602083 |
| USH1G | 7% | 17q24-q25 | USH1G | SANS | Proteinska skela | 461 | Q495M9 | OMIM: 606943 |
| USH2A | 80% | 1q41 | USH2A | Usherin | Transmembransko vezanje | 5202 | O75445 | OMIM: 276901 |
| USH2C | 15% | 5q14.3-q21.1 | GPR98 | VLGR1b | Vrlo veliki GPCR | 6307 | Q8WXG9 | OMIM: 605472 |
| USH2D | 5% | 9q32-q34 | DFNB31 | Virlin | PDZ-domenski protein | 907 | Q9P202 | OMIM: 611383 |
| USH3A | 100% | 3q21-q25 | CLRN1 | Clarin-1 | Oblikovanje sinapsi | 232 | P58418 | OMIM: 276902 |
Usherov sindrom nasljeđuje se u autosomno recesivno obilježje. Nekoliko gena je povezano sa Usherovim sindromom, koristeći analize veza u porodicama pacijenata (tabela 1) i DNK sekvenciranje identificiranih lokusa.[4][5] Mutacija bilo kojeg od ovih gena vjerovatno će rezultirati Usherovim sindromom.
Klinički podtipovi Usher I i II povezani su s mutacijama bilo kojeg od šest (USH1B - G), odnosno tri (USH2A, C-D ) gena, dok je samo jedan gen, USH3A , do sada povezan s Usherom III. Druga dva gena, USH1A i USH2B , u početku su bili povezani s Usherovim sindromom, ali USH2B nije provjeren, a USH1A je pogrešno utvrđen i ne postoji.[6] Research in this area is ongoing.
Primjenom tehnika interakcijske analize, može se pokazati da identificirani genski proizvodi međusobno komuniciraju u jednom ili više većih proteinskih kompleksa. Ako jedna od komponenti nedostaje, ovaj proteinski kompleks ne može ispuniti svoju funkciju u živoj ćeliji, a vjerojatno dolazi i do njegove degeneracije . Predloženo je da funkcija ovog proteinskog kompleksa učestvuje u transdukciji signala ili u adheziujama senzornih ćelija.[5]
Studija pokazuje da su tri proteina povezana sa genima Usherovog sindroma (PCDH15, CDH23, GPR98) takođe uključena u razvoj slušne kore, kod miša i makaka. Njihov nedostatak ekspresije inducira smanjenje broja parvalbuminskih interneurona. Pacijenti sa mutacijama ovih gena mogli bi imati oštećenja slušnog korteksa.[7]
Patofiziologija
[uredi | uredi izvor]Progresivno sljepilo Usherovog sindroma posljedica je bolesti retinitis pigmentosa. Fotoreceptorske ćelije obično počinju degenerirati iz vanjske periferije u središte mrežnjače, uključujući makulu. Degeneracija se obično prvo primijeti kao noćno sljepilo (niktalopija); periferni vid se postepeno gubi, ograničavajući vidno polje (tunelski vid, koje uglavnom napreduje do potpune sljepoće. Kvalifikator „pigmentoza“ odražava činjenicu da nakupine pigmenta, u naprednim fazama degeneracije, mogu biti vidljive pomoću oftalmoskopa.
Oštećenje sluha povezano s Usherovim sindromom uzrokovano je oštećenim trepljastim ćelijama k u pužnici unutrašnjeg uha koje sprečavaju električne impulse da dođu do mozga. To je oblik dysacusis.
Dijagnoza
[uredi | uredi izvor]Budući da je Usherov sindrom još uvijek neizlječiv, korisno je dijagnosticirati djecu prije nego što razviju karakteristično noćno sljepilo. Neke preliminarne studije sugeriraju da čak 10% urođeno gluhe djece može imati Usherov sindrom.[1] Međutim, pogrešna dijagnoza može imati loše posljedice.
Najjednostavniji pristup dijagnosticiranju Usherovog sindroma je ispitivanje karakterističnih hromosomskih mutacija. Alternativni pristup je elektroretinografija, iako je sa djeciom često neupotrebljiva, jer njegova nelagoda također može učiniti rezultate nepouzdanim.[1] Roditeljska krv je važan faktor u dijagnozi. Usherov sindrom Mogu biti indicirani ako je dijete duboko gluho od rođenja i posebno sporo hoda.
Trinaest drugih sindroma mogu pokazivati znakove slične Usherovom sindromu, kao što su Alportov sindrom, Alströmov sindrom, Bardet-Biedlov sindrom, Cockayneov sindrom, spondiloepifizna displazija kongenita, Flynn–Airdov sindrom, Friedreichova ataksija, Hurlerov sindrom (MPS-1), Kearns – Sayreov sindrom (CPEO), Norriejev sindrom, osteopetroza (Albers–Schonbergova bolest), Refsumova bolest (bolest skladištenja fitanske kiseline) i Zellwegerov sindrom (cerebrohepatorenalni sindrom).
Liječenje
[uredi | uredi izvor]Budući da je Usherov sindrom posljedica gubitka gena, genska terapija koja vraća odgovarajući protein ("zamjena gena") može ga ublažiti, pod uslovom da dodani protein postane funkcionalan. Nedavna istraživanja modela nokaut-miševa pokazala su da se jedan oblik bolesti – povezan sa mutacijom u miozinu VIIa – može ublažiti zamjenom mutantnog gena pomoću lentivirusa.[8] Međutim, neki mutirani geni povezani sa Usherovim sindromom kodiraju vrlo velike proteine – najvažnije, USH2A i GPR98' protein, koji imaju otprilike po 6.000 aminokiselinskih ostataka. Terapija zamjene gena za tako velike proteine može biti teška.
Epidemiologija
[uredi | uredi izvor]Usherov sindrom odgovoran je za većinu gluhoslijepoća.[9] U SAD, javlja se otprilike kod 1/23,000 ljudi[10] u norveškoj 1/28.000,[11] a u Njemačkoj 1/12.500.[12] People with Usher syndrome represent roughly one-sixth of people with retinitis pigmentosa.
Historija
[uredi | uredi izvor]Usherov sindrom je dobio ime po škotskom oftalmologu Charlesu Usheru, koji je 1914. godine na osnovu 69 slučajeva ispitivao patološko ispoljavanje i nasljeđivanje ove bolesti..[13] However, it was first described in 1858 by Albrecht von Gräfe, a pioneer of modern ophthalmology.[14] Izvijestio je o slučaju gluhog pacijenta sa retinitis pigmentosa, koji je imao dva brata s istim simptomima. Tri godine kasnije, jedan od njegovih učenika, Richard Liebreich, ispitivao je stanovništvo Berlina na oboljenja od gluhoće sa retinitis pigmentosa.[15] Liebreich je primijetio da je Usherov sindrom recesivan, jer su se slučajevi kombinacija slijepila i gluhoće javljali posebno kod braće i sestara u krvnim vezama ili u porodicama s pacijentima različitih generacija. Njegova zapažanja pružila su prve dokaze za povezani prenos sljepoće i gluhoće, jer na porodičnim stablima nije pronađen nijedan izolirani slučaj nijednog od njih.
Životinjski modeli ove ljudske bolesti (kao što su nokaut-miševi i zebrica) nedavno su razvijeni za proučavanje efekata ovih mutacija i za ispitivanje potencijalnih lijekova za Usherov sindrom.
Poznatije osobe
[uredi | uredi izvor]- Rebecca Alexander, psihoterapeut, autor, dobitnica nagrade Helen Keller za postignuća.
- Christine "Coco" Roschaert, direktor Nepalskog projrkta gluhosljepila.[16]
- Catherine Fischer napisala je autobiografiju odrastanja s Usherovim sindromom u Louisiani, pod naslovom Orhideja iz Bayoua.[17]
- Vendon Wright napisao je dvije knjige koje opisuju njegov život s Usherovim sindromom, "Bio sam slijep, ali sada vidim"[18] and Through my eyes.[19]
- Christian Markovic, slijepo-gluhi ilustrator i dizajner; Fuzzy Wuzzy Designs.[20]
- John Tracy, sin glumca Spencera Tracyija i imenjak doktora za bolesti usta u Klinike John Tracy.
- James D. Watson, suotkrivač DNK heliksa i nobelovac, ima homozigotnu mutaciju USH1B , prema njegovom objavljenom genomu.[21] Nije jasno zašto nije razvio sindrom. Ovaj nedostatak genetičke penetracije pokazuje da je ekspresija fenotipa Usherovog sindroma možda složenija nego što se prvobitno pretpostavljalo.
- Izraelski gluhonijemi glumački ansambl Nalaga'at (dodirnite) gluho-slijepog glumačkog ansambla sastoji se od 11 gluhonijemih glumaca, od kojih je većini dijagnosticiran Usherov sindrom. Pozorišna grupa izvela je nekoliko predstava i pojavila se lokalno u Izraelu i inostranstvu u Londonu i Broadwayu.[22]
- Katie Kelly, paraolimpijac koji je osvajio zlatnu medalju.
- Teigan Van Roosmalen, paraolimpijac.
- Cyril Axelrod, katolički svećenik.
- Rachel Chaikof, rana implantacija spužnicamwspužnice, primateljica i osnivačica potrala cochlearimplantonline.com.
- Robert Tarango, prva gluhoslijepa osoba koja je glumila u filmu, u ulozi Artija u kratkom filmu (Feeling Through), nominiranom za Oscara
Reference
[uredi | uredi izvor]- ^ a b c Mets MB, Young NM, Pass A, Lasky JB (2000). "Early diagnosis of Usher syndrome in children". Transactions of the American Ophthalmological Society. 98: 237–45. PMC 1298229. PMID 11190026.
- ^ Hope CI, Bundey S, Proops D, Fielder AR (1997). "Usher syndrome in the city of Birmingham — prevalence and clinical classification". British Journal of Ophthalmology. 81 (1): 46–53. doi:10.1136/bjo.81.1.46. PMC 1721995. PMID 9135408.
- ^ Roux AF, Faugere V, Le Guedard S, Pallares-Ruiz N, Vielle A, Chambert S, Marlin S, Hamel C, Gilbert B, Malcolm S, Claustres M (2006). "Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%". J Med Genet. 43 (9): 763–768. doi:10.1136/jmg.2006.041954. PMC 2564578. PMID 16679490.
Ouyang XM, Yan D, Du LL, Hejtmancik JF, Jacobson SG, Nance WE, Li AR, Angeli S, Kaiser M, Newton V, Brown SD, Balkany T, Liu XZ (2005). "Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population". Hum Genet. 116 (4): 292–299. doi:10.1007/s00439-004-1227-2. PMID 15660226. S2CID 22812718. - ^ Petit, C (2001). "Usher syndrome: from genetics to pathogenesis" (PDF). Annual Review of Genomics and Human Genetics. 2: 271–97. doi:10.1146/annurev.genom.2.1.271. PMID 11701652. S2CID 505750. Arhivirano s originala (PDF), 3. 5. 2019.
- ^ a b Reiners, J; Nagel-Wolfrum, K; Jürgens, K; Märker, T; Wolfrum, U (2006). "Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease" (PDF). Experimental Eye Research. 83 (1): 97–119. doi:10.1016/j.exer.2005.11.010. PMID 16545802. Arhivirano s originala (PDF), 3. 5. 2019.
- ^ Gerber, S; Bonneau, D; Gilbert, B; Munnich, A; Dufier, JL; Rozet, JM; Kaplan, J (2006). "USH1A: chronicle of a slow death". American Journal of Human Genetics. 78 (2): 357–9. doi:10.1086/500275. PMC 1380243. PMID 16400615.
- ^ Libé-Philippot, Baptiste; Michel, Vincent; Monvel, Jacques Boutet de; Gal, Sébastien Le; Dupont, Typhaine; Avan, Paul; Métin, Christine; Michalski, Nicolas; Petit, Christine (25. 7. 2017). "Auditory cortex interneuron development requires cadherins operating hair-cell mechanoelectrical transduction". Proceedings of the National Academy of Sciences (jezik: engleski). 114 (30): 7765–7774. doi:10.1073/pnas.1703408114. ISSN 0027-8424. PMC 5544301. PMID 28705869.
- ^ Hashimoto T, Gibbs D, Lillo C, Azarian SM, Legacki E, Zhang XM, Yang XJ, Williams DS (2007). "Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B". Gene Therapy. 14 (7): 584–594. doi:10.1038/sj.gt.3302897. PMID 17268537.
- ^ Vernon M (1969). "Usher's syndrome — deafness and progressive blindness. Clinical cases, prevention, theory and literature survey". Journal of Chronic Diseases. 22 (3): 133–151. doi:10.1016/0021-9681(69)90055-1. PMID 4897966.
- ^ Boughman J, Vernon M, Shaver K (1983). "Usher syndrome: Definition and estimate of prevalence from two high-risk populations". Journal of Chronic Diseases. 36 (8): 595–603. doi:10.1016/0021-9681(83)90147-9. PMID 6885960.
- ^ Grøndahl J (1987). "Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway". Clin. Genet. 31 (4): 255–264. doi:10.1111/j.1399-0004.1987.tb02804.x. PMID 3594933. S2CID 26853136.
- ^ Otterstedde CR, Spandau U, Blankenagel A, Kimberling WJ, Reisser C (2001). "A new clinical classification for Usher's syndrome based on a new subtype of Usher's syndrome type I". Laryngoscope. 111 (1): 84–86. doi:10.1097/00005537-200101000-00014. PMID 11192904.
- ^ Usher C (1914). "On the inheritance of Retinitis pigmentosa with notes of cases". Roy. Lond. Ophthalmol. Hosp. Rep. 19: 130–236.
- ^ von Gräfe A (1858). "Exceptionelles Verhalten des Gesichtsfeldes bei Pigmententartung der Netzhaut". Archiv für Ophthalmologie. 4: 250–253.
- ^ Liebreich R (1861). "Abkunft aus Ehen unter Blutsverwandten als Grund von Retinitis pigmentosa". Dtsch. Klin. 13: 53.
- ^ "Tactile The World". Tactile The World.
- ^ Carroll C, Fischer CH (2001). Orchid of the Bayou: A Deaf Woman Faces Blindness. Gallaudet University Press. ISBN 978-1-56368-104-2.
- ^ Wright V (2007). I was blind but now I can see. Authorhouse. ISBN 978-1-4208-9101-0.
- ^ Wright V (2007). Through my eyes. Pipers' Ash Ltd. ISBN 978-1-904494-86-7.
- ^ "Who's Fuzzy". Fuzzy Wuzzy Design. Arhivirano s originala, 29. 6. 2021. Pristupljeno 18. 6. 2021.
- ^ Green RC, Annas GJ (2008). "The Genetic Privacy of Presidential Candidates". New England Journal of Medicine. 359 (21): 2192–2193. doi:10.1056/NEJMp0808100. PMC 2925179. PMID 19020322.
- ^ "Arhivirana kopija". Arhivirano s originala, 24. 11. 2010. Pristupljeno 18. 6. 2021.CS1 održavanje: arhivirana kopija u naslovu (link)
Dopunska literatura
[uredi | uredi izvor]- Stiefel SH, Lewis RA (1991). The Madness of Usher's: Coping With Vision and Hearing Loss/Usher Syndrome Type II. Business of Living Publications. ISBN 978-1-879518-06-3.
- Duncan E, Prickett HT (1988). Usher's Syndrome: What It Is, How to Cope, and How to Help. Charles C. Thomas. ISBN 978-0-398-05481-6.
- Vernon M (1986). Answers to your questions about Usher's syndrome (retinitis pigmentosa with hearing loss). Foundation Fighting Blindness. ASIN B00071QLJ6.
- Vernon M (1969). Usher's syndrome: Deafness and progressive blindness : clinical cases, prevention, theory and literature survey. Pergamon Press. ASIN B0007JHOJ4.
Vanjski linkovi
[uredi | uredi izvor]- GeneReviews/NCBI/NIH/UW entry on Usher Syndrome Type I
- GeneReviews/NCBI/NIH/UW entry on Usher Syndrome Type II
- NCBI Genetic Testing Registry
- General overview from the NIH
- Usher Syndrome Information from the National Institute on Deafness and Other Communication Disorders (NIDCD).