Reazione SN1
La reazione SN1 è una reazione di sostituzione in chimica organica, il cui nome si riferisce al simbolo Hughes-Ingold del meccanismo. "SN" sta per sostituzione nucleofila e "1" indica che il passaggio che determina la velocità è unimolecolare[1][2]. Pertanto, l'equazione della velocità viene spesso mostrata come avente dipendenza di primo ordine dall'elettrofilo e dipendenza di ordine zero dal nucleofilo. Questa relazione vale per situazioni in cui la quantità di nucleofilo è molto maggiore di quella dell'intermedio. Invece, l'equazione della velocità può essere descritta in modo più accurato utilizzando la cinetica dello stato stazionario. La reazione coinvolge un intermedio carbocationico ed è comunemente osservata nelle reazioni di alogenuri alchilici secondari o terziari in condizioni fortemente basiche o, in condizioni fortemente acide, con alcoli secondari o terziari. Con alogenuri alchilici primari e secondari, si verifica in alternativa la reazione SN2. Nella chimica inorganica, la reazione SN1 è spesso nota come meccanismo dissociativo. Questa via di dissociazione è ben descritta dall'effetto cis. Un meccanismo di reazione è stato proposto per la prima volta da Christopher Ingold e suoi collaboratori nel 1940[3]. Questa reazione non dipende molto dalla forza del nucleofilo, a differenza del meccanismo SN2. Questo tipo di meccanismo prevede due passaggi: il primo passo è la ionizzazione dell'alogenuro alchilico in presenza di acetone acquoso o alcol etilico. Questo passaggio fornisce un carbocatione come intermedio.
Nella prima fase del meccanismo SN1 si forma un carbocatione, che è planare, e quindi l'attacco del nucleofilo (seconda fase) può avvenire da entrambi i lati per dare un prodotto racemico, ma in realtà non avviene la racemizzazione completa. Questo perché la specie nucleofila attacca il carbocatione anche prima che lo ione degli alogenuri in partenza si sia allontanato sufficientemente dal carbocatione. Lo ione alogenuro caricato negativamente protegge il carbocatione dall'attacco sul lato anteriore, e viene preferito l'attacco sul retro che porta all'inversione di configurazione. Quindi il prodotto reale è senza dubbio costituito da una miscela di enantiomeri, ma predominerebbero gli enantiomeri con configurazione invertita e non si verifica una racemizzazione completa.
Meccanismo
[modifica | modifica wikitesto]Un esempio di una reazione che avviene con un meccanismo di sostituzione SN1 è l'idrolisi del 2-bromo-2-metilpropano (bromuro di t-butile) che forma 2-metil-2-propanolo:

Questa reazione SN1 avviene in tre fasi:
- Formazione di un carbocatione t-butile mediante separazione di un gruppo uscente (un anione bromuro) dall'atomo di carbonio; questo passaggio è lento:[4]

- Attacco nucleofilo: il carbocatione reagisce con il nucleofilo. Se il nucleofilo è una molecola neutra (cioè un solvente) è necessario un terzo passaggio per completare la reazione. Quando il solvente è acqua, l'intermedio è uno ione ossonio. Questa fase di reazione è veloce.

- Deprotonazione: rimozione di un protone sul nucleofilo protonato mediante l'acqua, che agisce come base formando l'alcol e uno ione idronio. Questa fase di reazione è veloce.
Legge di velocità
[modifica | modifica wikitesto]Sebbene la legge di velocità della reazione SN1 sia spesso considerata di primo ordine negli alogenuri alchilici e di ordine zero nei nucleofili, questa è una semplificazione che vale solo in determinate condizioni. Sebbene sia anch'essa un'approssimazione, la legge di velocità derivata dall'approssimazione dello stato stazionario (SSA) fornisce maggiori informazioni sul comportamento cinetico della reazione SN1. Si consideri il seguente schema di reazione per il meccanismo mostrato sopra:

Sebbene sia un carbocatione terziario relativamente stabile, il catione t-butile è una specie ad alta energia che è presente a concentrazioni molto basse e non può essere osservata direttamente in condizioni normali. Pertanto, l'approssimazione dello stato stazionario può essere applicato a questa specie:
- Ipotesi allo stato stazionario:
- Concentrazione del catione t-butile, basata su ipotesi di stato stazionario:
- Velocità complessiva di reazione, ipotizzando un passaggio finale rapido:
- Legge del tasso stazionario, collegando (2) a (3):
In condizioni sintetiche normali, il nucleofilo entrante è più nucleofilo del gruppo uscente ed è presente in eccesso. Inoltre, gli esperimenti cinetici sono spesso condotti in condizioni di velocità iniziali (conversione dal 5 al 10%) e senza l'aggiunta di bromuro, quindi la concentrazione è trascurabile. Per questi motivi, spesso vale:
In queste condizioni, la legge sui tassi SSA si riduce alla velocità:
che è la legge sui tassi del primo ordine descritta nei libri di testo. In queste condizioni, la concentrazione del nucleofilo non influisce sulla velocità della reazione e la modifica del nucleofilo (ad esempio da H2O a MeOH) non influisce sulla velocità di reazione, sebbene il prodotto sia ovviamente diverso. In questo regime, la prima fase (ionizzazione dell'alchil bromuro) è lenta, determinante e irreversibile, mentre la seconda (addizione nucleofila) è veloce e cineticamente invisibile.
Tuttavia, in determinate condizioni, è possibile osservare cinetiche di reazione non del primo ordine. In particolare, quando è presente una grande concentrazione di bromuro mentre la concentrazione di acqua è limitata, diventa importante cineticamente l'inverso della prima fase. Come indica la legge sui tassi SSA, in queste condizioni esiste una dipendenza frazionaria (tra zero e primo ordine) dalla concentrazione [H2O], mentre esiste una dipendenza frazionaria negativa da [Br–]. Pertanto, si osserva spesso che le reazioni SN1 rallentano quando una fonte esogena del gruppo uscente (in questo caso il bromuro) viene aggiunta alla miscela di reazione. Questo è noto come effetto ionico comune e l'osservazione di questo effetto è la prova di un meccanismo SN1 (sebbene l'assenza di un effetto ionico comune non lo escluda)[5][6].
Scopo
[modifica | modifica wikitesto]Il meccanismo SN1 tende a dominare quando l'atomo di carbonio centrale è circondato da gruppi voluminosi perché tali gruppi ostacolano stericamente la reazione SN2. Il carbocatione risultante è anche stabilizzato sia dalla stabilizzazione induttiva che dall'iperconiugazione da gruppi alchilici attaccati. Il postulato di Hammond-Leffler suggerisce che anche questo aumenterà il tasso di formazione di carbocationi. Il meccanismo SN1 quindi domina nelle reazioni ai centri alchilici terziari.
Un esempio di una reazione che procede in modo SN1 è la sintesi del 2,5-dicloro-2,5-dimetilesano dal corrispondente diolo con acido cloridrico concentrato:[7]

All'aumentare delle sostituzioni alfa e beta rispetto ai gruppi uscenti, la reazione viene deviata da SN2 a SN1.
Stereochimica
[modifica | modifica wikitesto]L'intermedio carbocationico che si forma nella fase di determinazione della velocità della reazione (RDS) è un carbonio ibridato sp2 con geometria molecolare planare trigonale. Ciò consente due modi diversi per l'attacco nucleofilo, uno su ciascun lato della molecola planare. Se nessuna delle due vie è preferenzialmente favorita, queste due vie si verificano allo stesso modo, producendo una miscela racemica di enantiomeri se la reazione avviene in uno stereocentro[8]. Ciò è illustrato di seguito nella reazione SN1 di S-3-cloro-3-metilesano con uno ione ioduro, che produce una miscela racemica di 3-iodio-3-metilesano:
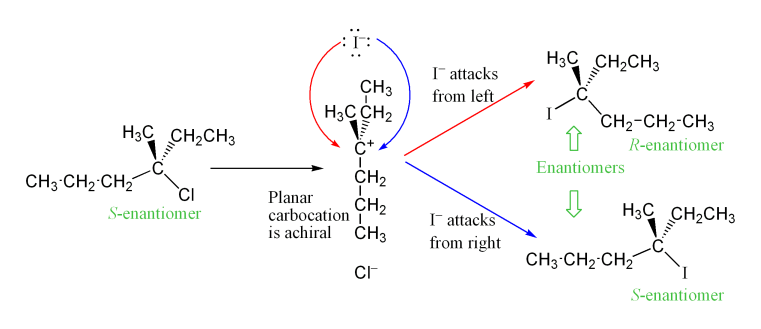
Tuttavia, si può osservare l'eccesso di uno stereoisomero, poiché il gruppo uscente può rimanere in prossimità dell'intermedio carbocationico per un breve periodo e bloccare l'attacco nucleofilo. Ciò è in contrasto con il meccanismo SN2, che è un meccanismo stereospecifico in cui la stereochimica è sempre invertita quando il nucleofilo entra dal lato posteriore del gruppo uscente.
Reazioni collaterali
[modifica | modifica wikitesto]Due reazioni collaterali comuni sono le reazioni di eliminazione e il riarrangiamento dei carbocationi. Se la reazione viene eseguita in condizioni di temperature alte o molto alte (che favoriscono un aumento dell'entropia), è probabile che predomini l'eliminazione E1 predomini, portando alla formazione di un alchene. A temperature più basse, le reazioni SN1 ed E1 sono reazioni competitive e diventa difficile favorire l'una rispetto all'altra. Anche se la reazione viene eseguita a freddo, si può formare un po' di alchene. Se si tenta di eseguire una reazione SN1 utilizzando un nucleofilo fortemente basico come lo ione idrossido o metossido, l'alchene verrà nuovamente formato, questa volta tramite un'eliminazione E2. Ciò sarà particolarmente vero se la reazione viene riscaldata. Infine, se l'intermedio carbocationico può riorganizzarsi in un carbocatione più stabile, darà un prodotto derivato dal carbocatione più stabile, piuttosto che dal semplice prodotto di sostituzione.
Effetti del solvente
[modifica | modifica wikitesto]Poiché la reazione SN1 comporta la formazione di un intermedio carbocationico instabile nella fase determinante la velocità (RDS), tutto ciò che può facilitare questo accelererà la reazione. I normali solventi di scelta scelti sono sia solventi polari (per stabilizzare gli intermedi ionici in generale) che protici (per solvatare il gruppo uscente in particolare). I tipici solventi protici polari includono acqua e alcoli, che agiranno anche come nucleofili e il processo è noto come solvolisi.
La scala Y correla i tassi di reazione di solvolisi di qualsiasi solvente (k) con quello di un solvente standard (80% v/v etanolo/acqua) (k0) attraverso la relazione:
dove una costante del reagente ( per il cloruro di t-butile) e un parametro del solvente[9]. Ad esempio, etanolo al 100% dà , etanolo al 50% in acqua dà e infine una concentrazione al 15% [10].
Note
[modifica | modifica wikitesto]- ^ (EN) L.G. Wade Jr., Organic Chemistry, 6ª ed., Upper Saddle River, Pearson/Prentice Hall, 2005.
- ^ (EN) J. March, Advanced Organic Chemistry, 4ª ed., New York, Wiley, 1992, ISBN 0-471-60180-2.
- ^ (EN) Bateman, L.C., Church, M.G., Hughes, E.D., Ingold, C.K. e Taher, N.A., 188. Mechanism of substitution at a saturated carbon atom. Part XXIII. A kinetic demonstration of the unimolecular solvolysis of alkyl halides. (Section E) a general discussion, in Journal of the Chemical Society (Resumed), 1940, p. 979, DOI:10.1039/JR9400000979.
- ^ (EN) Peters, K.S., Nature of Dynamic Processes Associated with the SN1 Reaction Mechanism, in Chem. Rev., vol. 107, n. 3, 2007, pp. 859–873, DOI:10.1021/cr068021k.
- ^ (EN) Anslyn, Eric V. e Dougherty, Dennis A., Modern physical organic chemistry, Mill Valley, California, University Science Books, 2006, pp. 638–639, ISBN 1-891389-31-9.
- ^ (EN) Lowry, Thomas H. e =Richardson, Kathleen Schueller, Mechanism and theory in organic chemistry, 3ª ed., New York, Harper & Row., 1987, pp. 330–331, ISBN 0-06-044084-8.
- ^ (EN) Carl E. Wagner e Pamela A. Marshall, Synthesis of 2,5-Dichloro-2,5-dimethylhexane by an SN1 Reaction, in J. Chem. Educ., vol. 87, n. 1, 2010, pp. 81–83, DOI:10.1021/ed8000057.
- ^ (EN) Sorrell, Thomas N., Organic Chemistry, 2ª ed., University Science Books, 2006.
- ^ (EN) Ernest Grunwald e S. Winstein, The Correlation of Solvolysis Rates, in J. Am. Chem. Soc., vol. 70, n. 2, 1948, p. 846, DOI:10.1021/ja01182a117.
- ^ (EN) Arnold H. Fainberg e S. Winstein, Correlation of Solvolysis Rates. III.1 t-Butyl Chloride in a Wide Range of Solvent Mixtures, in J. Am. Chem. Soc., vol. 78, n. 12, 1956, pp. 2770, DOI:10.1021/ja01593a033.
Voci correlate
[modifica | modifica wikitesto]Altri progetti
[modifica | modifica wikitesto]Wikimedia Commons contiene immagini o altri file su Reazione SN1