Unión neuromuscular
La unión neuromuscular, unión mioneural o sinapsis neuromuscular es una estructura celular especializada para el contacto entre el axón de una neurona motora y la membrana de una fibra muscular.[1] Las neuronas motoras transmiten el impulso nervioso a larga distancia, desde su cuerpo celular (o soma) hasta su membrana presináptica. El impulso que viaja por el axón llega hasta el extremo en el que se liberan vesículas repletas de acetilcolina, que es el neurotransmisor. La aceticolina liberada fuera de la neurona se une a los receptores de la membrana postsináptica de la fibra muscular. Debido a que la membrana postsináptica es eléctricamente excitable por la unión del neurotransmisor a los receptores, el impulso nervioso de la neurona motora logra transmitirse desde el extremo del axón hacia la fibra muscular.[2]
| Unión neuromuscular | ||
|---|---|---|
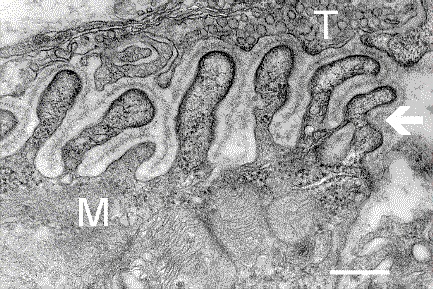 Micrografía electrónica que muestra una sección transversal de la unión neuromuscular. T es el axón terminal, M es la fibra muscular. La flecha muestra los pliegues de unión con la lámina basal. Las densidades postsinápticas pueden verse en las puntas entre los pliegues. La escala es de 0,3 micras. Fuente: NIMH | ||
 Vista detallada de la unión neuromuscular: 1. Terminal presináptica 2. Sarcolema 3. Vesícula sináptica 4. Receptor nicotínico 5. Mitocondria | ||
| Nombre y clasificación | ||
| Sinónimos |
| |
| Latín | synapsis neuromuscularis; junctio neuromuscularis | |
| TH | H2.00.06.1.02001 | |
| TH | H2.00.06.1.02001 | |
|
| ||
Estructura
editarLas fibras nerviosas motoras están canalizadas en los nervios desde su salida del sistema nervioso central. Las fibras nerviosas están constituidas por el axón, que transmite el impulso nervioso, y por las vainas de mielina que protegen al axón y están constituidas por células de Schwann. Con los nervios proyectándose hacía los músculos, la fibra nerviosa motora pierde la cobertura de mielina y se divide en pequeñas ramas axónicas cerca del área de las fibras musculares en el músculo, preparadas para recibir la sinapsis. Estos lugares en la fibra muscular son modificaciones de la membrana denominadas placas motoras, siendo una por cada fibra muscular. Sobre cada placa motora la rama axónica se ensancha para conformarse como un botón presináptico. La sinapsis entre el axón y la fibra muscular es protegida por una célula de Schwann, que aísla también la unión neuromuscular con el exterior.[3] En un corte transversal de la unión neuromuscular se distinguen tres partes:
- La membrana presináptica de la neurona motora o motoneurona. Especialización del axón para la acumulación y liberación del neurotransmisor cuando recibe el impulso nervioso. El axón con esta conformación se denomina axón terminal, aunque también recibe el nombre de botón presináptico o botón terminal. Este botón presináptico contiene multitud de mitocondrias y vesículas esféricas de 60 nanómetros de diámetro rellenas de acetilcolina como neurotransmisor.[4]
- Un espacio sináptico o hendidura sináptica. Es el espacio entre el botón presináptico y la membrana de la fibra muscular. La hendidura sináptica tiene entre 30 y 50 nanómetros de ancho. Entre ambas membranas se interpone una lámina basal que continua por los pliegues que forma la membrana postsináptica creando hendiduras secundarias.[4] La lámina basal en el espacio sináptico contiene proteínas estructurales como colágeno o laminina. Existen diferencias en la longitud de las hendiduras secundarias entre las fibras musculares lentas y rápidas.[4]
- Membrana postsináptica de la fibra muscular. Presenta la placa motora rica en proteínas receptoras de los neurotransmisores. En las uniones colinérgicas, que son las mediadas por la acetilcolina, se encuentran en una proporción de aproximadamente 104 (10 000, diez mil) receptores por cada μm2 en la placa motora. Igualmente se pueden encontrar en las hendiduras sinápticas secundarias y, aunque el número es menor, contribuyen a la acción de los neurotransmisores.[3][5] En la membrana basal de las hendiduras sinápticas también hay gran concentración de enzimas acetilcolinesterasas liberadas por la células musculares para degradar la acetilcolina unida a las membranas y permitir que la membrana muscular retorne al estado de reposo.[3]
T= (arriba) es el axón terminal repleto de vesículas.
M= es la fibra muscular.
La flecha blanca a la derecha muestra los pliegues de unión con la lámina basal.
Las densidades postsinápticas pueden verse en las puntas entre los pliegues. La escala es de 0,3 micras. Microscopio electrónico .
Las uniones neuromusculares de las fibras musculares lisas inervadas por fibras nerviosas del sistema nerviosos autónomo no presentan una estructura tan definida en las membranas pre y postsináptica, y su espacio sináptico es de menor diámetro. Además, a diferencia de los axones motores que liberan siempre acetilcolina, el neurotransmisor para las fibras nerviosas simpáticas es la noradrenalina y para las fibras nerviosas parasimpáticas la acetilcolina (colinérgica). En las uniones no colinérgicas de las fibras parasimpáticas los nucleósidos funcionan también como neurotransmisores.[4][6]
Fisiología de la unión neuromuscular
editarTransmisión del impulso eléctrico
editarLa membrana de la motoneurona es eléctricamente excitable y transmite el impulso nervioso desde el sistema nervioso central hasta el botón terminal. El impulso nervioso despolariza la membrana presináptica que activa los canales de calcio dependiente de voltaje. Esta activación permite la incorporación de grandes cantidades de iones de calcio al interior del axón terminal.[3][6]
Formación y liberación de acetilcolina
editarLas vesículas de aproximadamente 50 nanómetros (nm) de diámetro son generadas a partir del aparato de Golgi localizadas en el sistema nervioso central. La acetilcolina es sintetizada en el terminal del axón que son encapsuladas en las vesículas que contienen aproximadamente 10.000 moléculas de acetilcolina. Las vesículas son transportada a la membrana presináptica, en donde se llegan a acumular alrededor de 300 000 vesículas.[2][7] Cuando un impulso nervioso llega al botón presináptico, se abren los canales de calcio activados por voltaje que permite la incorporación de gran cantidad de iones calcio al interior. Estos iones atraen las vesículas a la membrana presináptica para fusionarse con la membrana terminal y liberando al espacio sináptico las moléculas de acetilcolina por exocitosis.[8]
Los receptores de acetilcolina están constituidos por cinco subunidades: dos subunidades alfa, y tres subunidades beta, delta y gamma. Se disponen de forma radial formando un canal que permanece cerrado hasta que dos moléculas de acetilcolina se unen a las subunidades alfa. Esta unión produce un cambio conformacional que abre el canal para el paso de iones de sodio al interior de la fibra muscular.[8] La incorporación de gran cantidad de iones de sodio causa la despolarización de la membrana postsináptica al generar un potencial positivo en la placa motora que inicia un potencial de acción. La propagación del potencial de acción a lo largo de la membrana de la fibra muscular desemboca en la contracción de la fibra muscular.[4][8]
Degradación de la acetilcolina y vuelta al estado de reposo
editarMientras moléculas de acetilcolina estén liberadas en el espacio sináptico se seguirán activando los receptores de acetilcolina. Para que la membrana de la placa muscular vuelva al estado de reposo el neurotransmisor se elimina por dos medios. Algunas moléculas difunden hacía el exterior del espacio sináptico donde ya no pueden participar en la despolarización de la membrana.[8] La mayor parte de la acetilcolina es degradada por la enzima acetilcolinesterasa, que es liberada por la fibra muscular. La enzima divide cada neurotransmisor en dos subunidades: acetato y colina. Las subunidades son reabsorbidas por el botón sináptico o se terminan de degradar en el espacio sináptico.[4] La catálisis es una de las más rápidas conocidas de manera que aproximadamente 5 moléculas de acetilcolina por milisegundo son degradadas por la enzima acetilcolinesterasa. La proporción de receptores de membrana acetilcolina es aproximadamente 5 a 10 veces mayor de enzima acetilcolinesterasa, lo que permite una interacción de 50 a 75% de toda la acetilcolina liberada con los receptores en la membrana postsináptica.[7]
Formación de las uniones durante el desarrollo
editarLos componentes del axón terminal y de la placa motora de la fibra muscular son sintetizadas e interaccionan entre sí para constituir las uniones neuromusculares durante el desarrollo embrionario. Los axones motores en desarrollo se extienden hacía los músculos en formación. Los conos axónicos de las neuronas llegan cuando los mioblastos se fusionan para constituir los miotubos. Los axones se dividen en varias ramas para dividir las diferentes fibras musculares en formación. Los conos axónicos minutos después hacer contacto con la membrana de la fibra muscular tienen capacidad de liberar acetilcolina con capacidad de transmitir el impulso nervioso.[9]
La unión primitiva, pese a ser débil, desencadena cambios en la membrana postsináptica. Los miotubos en formación para constituir las fibras musculares presentan receptores de acetilcolina de manera dispersa a lo largo de su membrana. La constitución de las sinapsis neuromusculares conduce a una concentración de los receptores de la acetilcolina en las placas musculares que se observan en las uniones maduras. En un día o dos el axón terminal y placa motora son estructuras diferenciables, y en una semana el botón terminal se ensancha para cubrir toda la placa motora, el citoesqueleto despeja el área que es rellena por multitud de mitocondrias y vesículas de acetilcolina. Otros componentes sintetizados por la fibra muscular como Agrin, MuSK o las proteínas de adhesión como las cadherinas participan en la constitución y estabilización de las uniones neuromusculares durante todo el proceso de sinaptogénesis.[9]
Patologías
editarExisten patologías relacionadas con la unión neuromuscular que generan dificultades motoras.
Enfermedades autoinmunes
editarMiastenia Gravis
editarMiastenia Gravis es una enfermedad autoinmune que afecta a la transmisión del impulso nervioso del axón terminal a la fibra muscular.[7] El sistema inmunitario del organismo ataca a los receptores de la acetilcolina de la membrana postsináptica de las fibra musculares. Como consecuencia no hay transmisión del impulso nervioso y la membrana muscular no se despolariza. La manifestación clínica es una progresiva debilidad muscular e incapacidad de realizar movimientos.[4][10][11] Afecta entre el 2 y 20 personas por cada millón de habitantes, con preferencia entre la población femenina y su desarrollo suele ser tardío en occidente, si bien la incidencia en juveniles es más frecuente entre la población asiática. La enfermedad se ha observado que es heredable en el 2% de los casos.[7][10] Actualmente no existe tratamiento para la cura completa de un cuadro de miastenia gravis pero si para mitigar los peores manifestaciones de la enfermedad. Uno de los tratamientos más comunes es el uso de inhibidores de la expresión de acetilcolinesterasa para compensar la pérdida de receptores de acetilcolina. En otro tipo de casos puede resultar efectivo la administración de esteroides o de inmunomoduladores que atacan a los anticuerpos que atacan a los componentes de la placa motora que provocan la enfermedad. En casos donde se observa un desarrollo anómalo del timo una extirpación parcial o completa del mismo puede significar una mejoría.[7][10]
La miastenia gravis fue descrita sistemáticamente por primera vez por Thomas Willis en el siglo XVII. El primer tratamiento con éxito de la enfermedad fue desarrollado por Mary Walker en la década de 1930 al identificar que una inhibición de la enzima acetilcolinesterasa causaba una mejoría en los pacientes. En los años 70 se descubre que la causa de la enfermedad es un ataque autoinmune de los receptores de acetilcolina en las placas motoras y se empiezan a determinar los primeros marcadores mediante serología para facilitar el diagnóstico de la enfermedad.[7]
Síndrome de Lambert-Eaton
editarEl síndrome Lambert-Eaton es una enfermedad autoinmune neurodegenerativa debido a una fusión anormal de las vesículas que contienen acetilcolina en la membrana presináptica. El sistema inmunitario ataca principalmente a las proteínas responsables de la liberación de los iones de calcio intracelular que sirve de señal para la el tráfico interno de las vesículas y su fusión con la membrana presináptica. Como consecuencia, la cantidad de moléculas de acetilcolina liberadas al espacio sináptico está reducido y la transmisión del impulso nerviosos se ve comprometida. Los primeros síntomas de la enfermedad es debilidad muscular, preferentemente en los miembros inferiores. A continuación se producen otras manifestaciones como perdida de reflejos y parálisis de los músculos inervados por los nervios craneales como los extraoculares. En estadios más avanzados también se observan afecciones propias del sistema nerviosos autónomo como sequedad en la boca, respuesta retardada del reflejo pupilar, estreñimiento, o ausencia de sudoración.[11][12] Este síndrome suele estar asociado a diversos cánceres de pulmón. También pueden estar asociados con otro tipo de tumores en páncreas y ovarios[4][11][12] Es una enfermedad que rara vez se presenta en niños que a su vez presenta una escasa incidencia en adultos de 1 caso cada 3,5 millones de personas. Un tercio de los pacientes son mujeres que presentan las primeras manifestaciones de la enfermedad a los 35 años, y aproximadamente los dos tercios restantes corresponden a varones hacía los 60 años de edad.[12]
La primera caracterización de la enfermedad fue realizada en la década de 1950.[11][12] Los tratamientos para combatir el síndrome Lambert-Eaton suelen perseguir, en primer lugar, la cura del cáncer que suele venir asociado a la enfermedad, con la esperanza de que la remisión del tumor ayude también a la recuperación del paciente del trastorno autoinmune. En los casos que esto no es posible, suele prepararse un tratamiento individualizado que incluyen la administración de inmunoglobulinas y otras drogas que han demostrado su eficacia, como la guanidina.[11][12] En otros casos, la transfusión periódica de plasma sanguíneo permite una mejora en el paciente al observarse una mejora de la transmisión del potencial nervioso. Sin embargo, es un tratamiento que no se ha generalizado y solo se ha probado a nivel experimental.[12]
Síndromes miasténicos congénitos
editarComprenden un conjunto de enfermedades congénitas relacionadas con la expresión deficiente de alguno de los componentes de la unión neuromuscular. Las deficiencias más comunes suelen aparecer al nacer o en los primeros años de vida, mientras que las patologías que aparecen en la edad adulta se asocian con genotipos específicos.[12] El conjunto total de estos síndromes es de una incidencia baja de menos de 4 enfermos por cada millón de habitantes para los al menos 18 síndromes identificados en relación con 17 genotipos diferentes. Las patologías se suelen clasificar de acuerdo a la parte de la unión neuromuscular afectada para facilitar el diagnóstico y tratamiento.[12]
Síndromes presinápticos. Representan un 5% de todos los casos documentados. Suelen estar relacionados con una producción deficiente de acetilcolina o su liberación por las vesículas presinápticas.[11][12]
Síndromes sinápticos. Comprenden mutaciones en las proteínas componentes de la membrana basal o relacionado con la liberación de la enzima acetilcolinesterasa al espacio sináptico. Comprenden alrededor del 15% de todos los síndromes miasténicos congénitos.[12]
Síndromes postsinápticos. Representan la mayoría de casos debido a la mayor proporción de proteínas que participan en la membrana postsináptica. Algunas de las mutaciones se relacionan con el proceso de concentración de los receptores de acetilcolina durante el desarrollo embrionario como el gen que codifica para la proteína Agrin u otros genes que igualmente codifican proteínas esenciales para la formación de la placa motora como MuSK, LRP4, Dok-7 o rapsyn. Otro conjunto de síndromes postsináptico están relacionados con una deficiencia en alguna de las subunidades que conforman los receptores de acetilcolina en la membrana postsináptica. De acuerdo a la subunidad afectada la patología puede ser más o menos benigna para el paciente.[11][12]
Toxinas que afectan a la transmisión del impulso nervioso
editarGarrapatas
editarLas garrapatas son los vectores de varias enfermedades. La saliva de las garrapatas de la familia de Ixodidae (garrapatas de coraza dura) es una toxina que puede causar parálisis muscular en algunas especies aparte del ser humano como perros y gatos. Varios días tras la mordedura el paciente desarrolla los primeros síntomas en forma de irritabilidad, somnolencia, diarrea y fiebre. Menos de 48 horas después se manifiesta debilidad muscular y parálisis progresiva en los miembros. Una vez retirada la garrapata los síntomas remiten en unos pocos días. Es una patología endémica del norte de América y Oceanía, aunque las especies de garrapatas australianas pueden desarrollar una variante más severa. Se desconoce el mecanismo exacto aunque se asume que la acción de la toxina bloquea la liberación de acetilcolina de los axones terminales.[12]
Otros artrópodos
editarOtros arácnidos producen toxinas que afectan dramáticamente la transmisión del impulso nervioso. El veneno de la viuda negra contiene una neurotoxina que estimula la liberación incontrolada de neurotransmisores como la adrenalina, dopamina y acetilcolina que vacían las vesículas en la membrana presináptica de los terminales axónicos en el sistema nervioso central y periférico.[12] El primer síntoma suele ser un dolor intenso en la zona de la mordedura. Con la toxina afectando a la internación autónoma de los vasos sanguíneos se observa una vasoconstricción, seguido de una rigidez muscular provocado por la hiperactividad de la acetilcolina liberada que se va extendiendo hacía el cuerpo. Con el tiempo se experimenta espasmos en la pared del abdomen, dolor de cabeza y dificultad para respirar por una bronconstricción. En los casos más extremos se puede llegar a observar una miocarditis, o inflamación del tejido muscular cardiaco, que puede llegar a ser fatal.[12]
El veneno de diversas especies de escorpiones también provocan una hiperliberación de las vesículas repletas de acetilcolina en el terminal axónico. Tras el pinchazo el paciente experimenta debilidad muscular acompañada de hipertensión arterial, arritmias cardíacas, miocarditis e incluso edema pulmonar.[12]
Veneno de serpientes
editarDiversas especies de serpientes elápidos que comprenden especies como cobras, mambas o serpientes de coral producen neurotoxinas especializadas en el bloqueo de la transmisión del impulso nervioso. De acuerdo a la toxina producida algunos venenos inhiben la transmisión presinápticamente, mientras que otros venenos actúan en una inhibición postsináptica del impulso nervioso.[12] Los primeros síntomas comienzan a los pocos minutos aunque pueden retrasarse a las 12 horas dependiendo de la especie. Las primeras manifestaciones clínicas son debilidad en los miembros acompañado de dolor muscular y náuseas. El cierre del párpado es frecuente y la hipersalivación puede o no ocurrir de acuerdo al tipo de veneno. El bloqueo de la transmisión del impulso nervioso es una característica propia del veneno de los elápidos y, aunque no atraviesen la barrera hematoencefálica, otras manifestaciones de sus toxinas pueden afectar a otras áreas sistema nervioso como bajada de la tensión arterial e hipoxia. Si no recibe atención médica adecuada la muerte acontece antes de las 48 horas, aunque puede ser antes dependiendo de la especie.[12]
El veneno de las serpientes marinas comparte muchas de las características de las serpientes terrestres al pertenecer a la misma familia Elapidae.[12]
Microorganismos
editarLa toxina botulínica liberada por la bacteria Clostridium botulinum actúa en la membrana presináptica impidiendo la liberación de la acetilcolina y provocando la destrucción del terminal del axón que puede regenerarse para volver a hacer sinapsis en la misma placa motora. Aproximadamente son identificados unos 1000 casos anuales alrededor del mundo, la mayoría entre la población infantil, por consumir aguas contaminadas.[10] Las bacterias se alojan en el sistema digestivo liberando toxinas cuyas primeras manifestaciones son disminución del tono muscular, ausencia de reflejos, dificultad en la respiración, estreñimiento y actividad anormal en los músculos internados por los nervios craneales. En los casos más extremos el paciente puede experimentar debilidad y parálisis descendente desde los músculos de cabeza y cuello hacía los miembros. La dificultad respiratoria puede agravarse hasta llegar a provocar la muerte. El tratamiento por intoxicación de Clostridium botulinum es la administración de antitoxinas.[4][10] La toxina botulínica aislada, por otra parte, se le ha encontrado uso en clínica y en ciertos tratamientos de belleza.[4]
Intoxicación por compuestos organofosfatos y carbamatos
editarAlgunos de los insecticidas usados en agricultura contienen en su composición organofosfatos y carbamatos. En forma de plaguicidas estos compuestos se encuentran en baja concentración para la eliminación de los insectos sin afectar a los seres humanos. Sin embargo, estos compuestos también se pueden utilizar a altas concentraciones como venenos o siendo los constituyentes de diversas armas biológicas. En menos de 24 horas tras la exposición el paciente comienza un cuadro de aumento de saliva, lagrimeo, incontinencia y defecación acompañados de otros síntomas como respiración arrítmica, parálisis de los miembros y delirios. Una parada respiratoria por un debilitamiento fatal en los músculos del diafragma e intercostales suele ser la causa de la muerte en los casos de envenenamiento extremo. Los organofosfatos y carbamatos inhiben de manera total o temporal las enzimas acetilcolinesterasas provocando una hiperactividad en la transmisión del impulso nervioso a la membrana postsináptica de la fibra muscular.[12]
Referencias
editar- ↑ OMS,OPS,BIREME (ed.). «Unión neuromuscular». Descriptores en Ciencias de la Salud, Biblioteca Virtual en Salud.
- ↑ a b Mancall, Elliott L. (2011). «Chapter 2. Overview of the Microstructure of the Nervous System». En David G. Brock, ed. Gray´s Clinical Neuroanatomy (en inglés). Elsevier. pp. 11-31. ISBN 978-1-4160-4705-6.
- ↑ a b c d Greenstein, Ben; Greenstein, Adam (2000). «Cellular Structures». Color Atlas of Neuroscience (en inglés). New York: Thieme. pp. 72-131. ISBN 0-86577-710-1.
- ↑ a b c d e f g h i j Elliott L. Mancall, David G. Brock, ed. (2011). «Chapter 22. Neuromuscular Junction». Gray´s Clinical Neuroanatomy (en inglés). Filadelfia, EE.UU.: Elsevier. pp. 377-380. ISBN 978-1-4160-4705-6.
- ↑ Abdillahi Omar et.al. (2020). «Physiology, Neuromuscular Junction.» [Fisiología, unión neuromuscular.] (en inglés.).
- ↑ a b Snell, Richard S (2014). «3. Fibras nerviosas, nervios periféricos, terminaciones receptoras y efectoras, dermatomas y actividad muscular». Neuroanatomía Clínica (7a edición). Barcelona (España): Wolkers Klwer. pp. 140-241. ISBN 978-0-7817-9427-5.
- ↑ a b c d e f Amato, Anthony A.; Russell, James A (2016). «25. Autoimmune Myasthenia Gravis». Neuromuscular Disorders (en inglés) (2ª edición). Nueva York (EE.UU): McGraw-Hill. pp. 581-619. ISBN 978-0-07-175462-0.
- ↑ a b c d Guyton, Arthur C.; Hall, John E. (2011). «7. Excitación del músculo esquelético: transmisión neuromuscular y acoplamiento excitación-contracción». En Elsevier, ed. Guyton y Hall Tratado de Fisiología Médica (12 edición). Barcelona (España). pp. 83-89. ISBN 978-84-8086-819-8.
- ↑ a b Patton, Bruce; Burgess, Robert W. (2005). «Chapter 10. Synaptogenesis». Developmental Neurobiology (en inglés) (4th edición). New York: Kluwer Academic. pp. 269-316. ISBN 0-306-48330-0.
- ↑ a b c d e Toy, Eugene C.; Simpson, Ericka; Pleitez, Milvia; Rosenfield, David; Tintner, Ron (2008). Case Files Neurology (en inglés). Nueva York, EE.UU.: McGraw-Hill. doi:10.1036/0071482873.
- ↑ a b c d e f g James S. Nelson, Hernando Mena, Joseph E Parisi, Sydney S. Schochet Jr, ed. (2003). «23. Neuromuscular diseases». Principles and Practice of Neuropathology (en inglés) (2ª edición). Nueva York, EE.UU.: Oxford University Press. pp. 524-577.
- ↑ a b c d e f g h i j k l m n ñ o p q r Amato, Anthony A; Russell, James A (2016). «26. Other Disorders of Neuromuscular Transmission». Neuromuscular Disorders (en inglés) (2ª edición). Nueva York (EE.UU): McGraw-Hill. pp. 620-655. ISBN 978-0-07-175462-0.
Datos: Q776995
Multimedia: Neuromuscular junction / Q776995