Cinasa

En bioquímica, una cinasa[2] o quinasa[2] es una enzima que cataliza la transferencia de grupos fosfato de moléculas donantes de fosfato de alta energía a sustratos específicos. Este proceso se conoce como fosforilación, donde el sustrato gana un grupo fosfato y la molécula de ATP de alta energía dona un grupo fosfato. Esta transesterificación produce un sustrato fosforilado y ADP. A la inversa, se denomina desfosforilación cuando el sustrato fosforilado dona un grupo fosfato y el ADP gana un grupo fosfato (produciendo un sustrato desfosforilado y la molécula de ATP de alta energía). Estos dos procesos, fosforilación y desfosforilación, ocurren cuatro veces durante la glucólisis.[3][4][5]
Las cinasas son parte de la familia más grande de fosfotransferasas. Las cinasas no deben confundirse con las fosforilasas, que catalizan la adición de grupos fosfato inorgánicos a un aceptor, ni con las fosfatasas, que eliminan los grupos fosfato (desfosforilación). El estado de fosforilación de una molécula, ya sea una proteína, un lípido o un carbohidrato, puede afectar su actividad, reactividad y su capacidad para unirse a otras moléculas. Por lo tanto, las cinasas son críticas en el metabolismo, la señalización celular, la regulación de proteínas, el transporte celular, la secreción y muchas otras vías celulares, lo que las hace muy importantes para el funcionamiento del ser humano y los demás seres vivos.
Bioquímica e importancia funcional
[editar]
Las cinasas median la transferencia de un resto fosfato de una molécula de alta energía (como el ATP) a su molécula de sustrato, como se ve en la figura siguiente. Se necesitan cinasas para estabilizar esta reacción porque el enlace fosfoanhídrido contiene un alto nivel de energía. Las cinasas orientan adecuadamente su sustrato y el grupo fosforilo dentro de sus sitios activos, lo que aumenta la velocidad de la reacción. Además, comúnmente usan residuos de aminoácidos cargados positivamente, que estabilizan electrostáticamente el estado de transición al interactuar con los grupos fosfato cargados negativamente. Alternativamente, algunas cinasas utilizan cofactores metálicos unidos en sus sitios activos para coordinar los grupos fosfato. Las proteínas cinasas pueden clasificarse como catalíticamente activas (canónicas) o como pseudocinasas, lo que refleja la pérdida evolutiva de uno o más de los aminoácidos catalíticos que posicionan o hidrolizan el ATP.[6] Sin embargo, en términos de resultados de señalización y relevancia de la enfermedad, tanto las cinasas como las pseudocinasas son importantes moduladores de señalización en las células humanas, lo que hace que las cinasas sean objetivos de fármacos muy importantes.[7]
Las cinasas se utilizan ampliamente para transmitir señales y regular procesos complejos en las células. La fosforilación de moléculas puede mejorar o inhibir su actividad y modular su capacidad para interactuar con otras moléculas. La adición y eliminación de grupos fosforilo proporciona a la célula un medio de control porque varias cinasas pueden responder a diferentes condiciones o señales. Las mutaciones en las cinasas que conducen a una pérdida o ganancia de función pueden causar cáncer[8] y enfermedades en humanos, incluidos ciertos tipos de leucemia y neuroblastomas, glioblastoma,[9] ataxia espinocerebelosa (tipo 14), formas de hipogammaglobulinemia, y muchos otros.[10]
Historia y clasificación
[editar]La primera proteína en ser reconocida como catalizadora de la fosforilación de otra proteína usando ATP fue observada en 1954 por Gene Kennedy, momento en el que describió una enzima hepática que catalizaba la fosforilación de caseína. En 1956, Edmond H. Fischer y Edwin G. Krebs descubrieron que la interconversión entre la fosforilasa ay la fosforilasa b estaba mediada por la fosforilación y la desfosforilación.[11] La cinasa que transfirió un grupo fosforilo a fosforilasa b, convirtiéndolo en fosforilasa a, se denominó fosforilasa cinasa. Años más tarde, se identificó el primer ejemplo de una cascada de cinasas, mediante la cual la proteína cinasa A (PKA) fosforila la fosforilasa cinasa. Al mismo tiempo, se encontró que la PKA inhibía la glucógeno sintasa, que fue el primer ejemplo de un evento de fosforilación que resultó en inhibición. En 1969, Lester Reed descubrió que la piruvato deshidrogenasa se inactivaba por fosforilación, y este descubrimiento fue la primera pista de que la fosforilación podría servir como un medio de regulación en otras vías metabólicas además del metabolismo del glucógeno. Ese mismo año, Tom Langan descubrió que la PKA fosforila la histona H1, lo que sugiere que la fosforilación podría regular las proteínas no enzimáticas. La década de 1970 incluyó el descubrimiento de proteínas cinasas dependientes de calmodulina y el descubrimiento de que las proteínas pueden fosforilarse en más de un residuo de aminoácido. La década de 1990 puede describirse como la "década de las cascadas de proteína cinasa". Durante este tiempo, se descubrieron la vía MAPK / ERK, las cinasas Jano (una familia de proteínas tirosina cinasas) y la cascada de cinasas dependiente de PIP3.[12]
Las cinasas se clasifican en grupos amplios según el sustrato sobre el que actúan: proteína cinasas, lípido cinasas, carbohidrato cinasas. Las cinasas se pueden encontrar en una variedad de especies, desde bacterias hasta moho, gusanos y mamíferos.[13] Se han identificado más de quinientas cinasas diferentes en humanos.[3] Su diversidad y su papel en la señalización los convierte en un interesante objeto de estudio. Varias otras cinasas actúan sobre moléculas pequeñas como lípidos, carbohidratos, aminoácidos y nucleótidos, ya sea para señalizarlas o para prepararlas para vías metabólicas. Las cinasas específicas a menudo reciben el nombre de sus sustratos. Las proteínas cinasas a menudo tienen múltiples sustratos y las proteínas pueden servir como sustratos para más de una cinasa específica. Por esta razón, las proteínas cinasas se nombran en función de lo que regula su actividad (es decir, Proteínas cinasas dependientes de calculina). A veces, se subdividen en categorías porque hay varias formas isoenzimáticas. Por ejemplo, las proteínas cinasas dependientes de AMP cíclico de tipo I y tipo II tienen subunidades catalíticas idénticas, pero diferentes subunidades reguladoras que se unen al AMP cíclico.[14]
Proteína cinasas
[editar]Las proteínas cinasas actúan sobre las proteínas, fosforilándolas en sus residuos de serina, treonina, tirosina o histidina. La fosforilación puede modificar la función de una proteína de muchas formas. Puede aumentar o disminuir la actividad de una proteína, estabilizarla o marcarla para su destrucción, localizarla dentro de un compartimento celular específico y puede iniciar o interrumpir su interacción con otras proteínas. Las proteína cinasas constituyen la mayoría de todas las cinasas y se estudian ampliamente.[15] Estas cinasas, junto con las fosfatasas, desempeñan un papel importante en la regulación de proteínas y enzimas, así como en la señalización celular.
Un punto común de confusión surge al pensar en las diferentes formas en que una célula logra la regulación biológica. Hay innumerables ejemplos de modificaciones covalentes que pueden sufrir las proteínas celulares; sin embargo, la fosforilación es una de las pocas modificaciones covalentes reversibles. Esto proporcionó el fundamento de que la fosforilación de proteínas es reguladora. El potencial para regular la función de la proteína es enorme dado que hay muchas formas de modificar covalentemente una proteína además de la regulación proporcionada por el control alostérico. En su Hopkins Memorial Lecture, Edwin Krebs afirmó que el control alostérico evolucionó para responder a las señales que surgen del interior de la célula, mientras que la fosforilación evolucionó para responder a las señales fuera de la célula. Esta idea es consistente con el hecho de que la fosforilación de proteínas ocurre con mucha más frecuencia en las células eucariotas en comparación con las células procariotas porque el tipo de célula más complejo evolucionó para responder a una gama más amplia de señales.[14]
Cinasas dependientes de ciclina
[editar]Las cinasas dependientes de ciclina (CDK) son un grupo de varias cinasas diferentes involucradas en la regulación del ciclo celular. Fosforilan otras proteínas en sus residuos de serina o treonina, pero las CDK deben unirse primero a una ciclina para ser activas.[16] Diferentes combinaciones de ciclinas y CDK específicas marcan diferentes partes del ciclo celular. Además, el estado de fosforilación de las CDK también es fundamental para su actividad, ya que están sujetas a la regulación de otras cinasas (como la cinasa activadora de CDK) y fosfatasas (como Cdc25).[17] Una vez que las CDK están activas, fosforilan otras proteínas para cambiar su actividad, lo que conduce a eventos necesarios para la siguiente etapa del ciclo celular. Si bien son más conocidas por su función en el control del ciclo celular, las CDK también desempeñan funciones en la transcripción, el metabolismo y otros eventos celulares.[18]
Debido a su papel clave en el control de la división celular, las mutaciones en las CDK a menudo se encuentran en las células cancerosas. Estas mutaciones conducen a un crecimiento descontrolado de las células, donde rápidamente pasan todo el ciclo celular de forma repetida.[19] Las mutaciones de CDK se pueden encontrar en linfomas, cáncer de mama, tumores de páncreas y cáncer de pulmón. Por lo tanto, se han desarrollado inhibidores de CDK como tratamientos para algunos tipos de cáncer.[19]
Proteína cinasas activadas por mitógenos
[editar]Las MAP cinasas (MAPK) son una familia de serina / treonina cinasas que responden a una variedad de señales de crecimiento extracelular. Por ejemplo, la hormona del crecimiento, el factor de crecimiento epidérmico, el factor de crecimiento derivado de las plaquetas y la insulina se consideran estímulos mitogénicos que pueden activar la vía MAPK. La activación de esta vía a nivel del receptor inicia una cascada de señalización mediante la cual Ras GTPasa intercambia GDP por GTP. A continuación, Ras activa la cinasa Raf (también conocida como MAPKKK), que activa MEK (MAPKK). MEK activa MAPK (también conocido como ERK), que puede regular la transcripción y la traducción. Mientras que RAF y MAPK son serina / treonina cinasas, MAPKK es una tirosina / treonina cinasa.
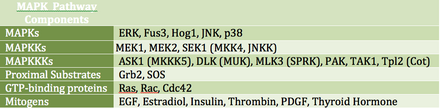
MAPK puede regular los factores de transcripción directa o indirectamente. Sus principales objetivos transcripcionales incluyen ATF-2, Chop, c-Jun, c-Myc, DPC4, Elk-1, Ets1, Max, MEF2C, NFAT4, Sap1a, STATs, Tal, p53, CREB y Myc. MAPK también puede regular la traducción mediante la fosforilación de la cinasa S6 en la subunidad mayor del ribosoma. También puede fosforilar componentes vía arriba en la cascada de señalización de MAPK, incluidos Ras, Sos y el propio receptor de EGF.[20]
El potencial carcinogénico de la vía MAPK la hace clínicamente significativa. Está implicado en procesos celulares que pueden conducir a un crecimiento descontrolado y la consiguiente formación de tumores. Las mutaciones dentro de esta vía alteran sus efectos reguladores sobre la diferenciación celular, la proliferación, la supervivencia y la apoptosis, todos los cuales están implicados en diversas formas de cáncer.[20]
Cinasas de lípidos
[editar]Las lípido cinasas fosforilan los lípidos en la célula, tanto en la membrana plasmática como en las membranas de los orgánulos. La adición de grupos fosfato puede cambiar la reactividad y localización del lípido y puede usarse en la transmisión de señales.
Fosfatidilinositol cinasas
[editar]
Las fosfatidilinositol cinasas fosforilan especies de fosfatidilinositol, para crear especies como fosfatidilinositol 3,4-bisfosfato (PI (3,4) P 2), fosfatidilinositol 3,4,5-trifosfato (PIP3) y fosfatidilinositol 3-fosfato (PI3P). Las cinasas incluyen fosfoinositido 3-cinasa (PI3K), fosfatidilinositol-4-fosfato 3-cinasa y fosfatidilinositol-4,5-bisfosfato 3-cinasa. El estado de fosforilación del fosfatidilinositol juega un papel importante en la señalización celular, como en la vía de señalización de la insulina, y también tiene un papel en la endocitosis, exocitosis y otros eventos de tráfico.[21][22] Las mutaciones en estas cinasas, como PI3K, pueden provocar cáncer o resistencia a la insulina.[23]
Las enzimas cinasas aumentan la velocidad de las reacciones al hacer que el grupo hidroxilo del inositol sea más nucleófilo, a menudo usando la cadena lateral de un residuo de aminoácido para actuar como base general y desprotonar el hidroxilo, como se ve en el mecanismo a continuación.[24] Aquí, se coordina una reacción entre el trifosfato de adenosina (ATP) y el fosfatidilinositol. El resultado final es un fosfatidilinositol-3-fosfato y un difosfato de adenosina (ADP). Las enzimas también pueden ayudar a orientar adecuadamente la molécula de ATP, así como el grupo inositol, para que la reacción avance más rápido. Los iones metálicos a menudo se coordinan para este propósito.[24]

Esfingosina cinasas
[editar]La esfingosina cinasa (SK) es una lípido cinasa que cataliza la conversión de esfingosina en esfingosina-1-fosfato (S1P). Los esfingolípidos son lípidos de membrana ubicuos. Tras la activación, la esfingosina cinasa migra desde el citosol a la membrana plasmática donde transfiere un fosfato γ (que es el último fosfato o el fosfato terminal) del ATP o GTP a la esfingosina. El receptor S1P es un receptor GPCR, por lo que S1P tiene la capacidad de regular la señalización de la proteína G. La señal resultante puede activar efectores intracelulares como ERK, Rho GTPasa, Rac GTPase, PLC y AKT / PI3K. También puede ejercer su efecto sobre las moléculas diana dentro de la célula. Se ha demostrado que S1P inhibe directamente la actividad histona desacetilasa de las HDAC. Por el contrario, la esfingosina desfosforilada promueve la apoptosis celular y, por lo tanto, es fundamental comprender la regulación de las SK debido a su papel en la determinación del destino celular. Investigaciones anteriores muestran que las SK pueden mantener el crecimiento de las células cancerosas porque promueven la proliferación celular, y SK1 (un tipo específico de SK) está presente en concentraciones más altas en ciertos tipos de cánceres.
Hay dos cinasas presentes en las células de mamíferos, SK1 y SK2. SK1 es más específico en comparación con SK2, y sus patrones de expresión también difieren. SK1 se expresa en células de pulmón, bazo y leucocitos, mientras que SK2 se expresa en células de riñón e hígado. La participación de estas dos cinasas en la supervivencia, proliferación, diferenciación e inflamación celular las convierte en candidatas viables para terapias quimioterapéuticas.[25]
Cinasas de carbohidratos
[editar]
Para muchos mamíferos, los carbohidratos proporcionan una gran parte del requerimiento calórico diario. Para recolectar energía de los oligosacáridos, primero deben descomponerse en monosacáridos para que puedan ingresar al metabolismo. Las cinasas juegan un papel importante en casi todas las vías metabólicas. La figura de la izquierda muestra la segunda fase de la glucólisis, que contiene dos reacciones importantes catalizadas por cinasas. El enlace anhídrido en 1,3 bisfosfoglicerato es inestable y tiene una alta energía. La 1,3-bisfosfoglicerato cinasa requiere ADP para llevar a cabo su reacción produciendo 3-fosfoglicerato y ATP. En el paso final de la glucólisis, la piruvato cinasa transfiere un grupo fosforilo del fosfoenolpiruvato al ADP, generando ATP y piruvato.
La hexocinasa es la enzima más común que utiliza la glucosa cuando ingresa por primera vez a la célula. Convierte D-glucosa en glucosa-6-fosfato transfiriendo el gamma fosfato de un ATP a la posición C6. Este es un paso importante en la glucólisis porque atrapa la glucosa dentro de la célula debido a la carga negativa. En su forma desfosforilada, la glucosa puede moverse hacia adelante y hacia atrás a través de la membrana con mucha facilidad.[26] Las mutaciones en el gen de la hexocinasa pueden conducir a una deficiencia de la hexocinasa que puede causar anemia hemolítica no esferocítica.[27]
La fosfofructocinasa, o PFK (phosphofructokinase), cataliza la conversión de fructosa-6-fosfato en fructosa-1,6-bisfosfato y es un punto importante en la regulación de la glucólisis. Los altos niveles de ATP, H+ y citrato inhiben la PFK. Si los niveles de citrato son altos, significa que la glucólisis está funcionando a un ritmo óptimo. Los altos niveles de AMP estimulan la PFK. La enfermedad de Tarui, una enfermedad de almacenamiento de glucógeno que conduce a la intolerancia al ejercicio, se debe a una mutación en el gen PFK que reduce su actividad.[28]
Otras cinasas
[editar]
Las cinasas actúan sobre muchas otras moléculas además de las proteínas, los lípidos y los carbohidratos. Hay muchos que actúan sobre los nucleótidos (ADN y ARN), incluidos los involucrados en la interconversión de nucleótidos, como las nucleósido fosfato cinasas y las nucleósido difosfato cinasas.[30] Otras moléculas pequeñas que son sustratos de cinasas incluyen creatina, fosfoglicerato, riboflavina, dihidroxiacetona, shikimato y muchas otras.
Riboflavina cinasa
[editar]La riboflavina cinasa cataliza la fosforilación de la riboflavina para crear flavina mononucleótido (FMN). Tiene un mecanismo de unión ordenado en el que la riboflavina debe unirse a la cinasa antes de que se una a la molécula de ATP.[31] Los cationes divalentes ayudan a coordinar el nucleótido.[31] El mecanismo general se muestra en la figura siguiente.

La riboflavina cinasa juega un papel importante en las células, ya que FMN es un cofactor importante. FMN también es un precursor del dinucleótido de flavina y adenina (FAD), un cofactor redox utilizado por muchas enzimas, incluidas muchas en el metabolismo. De hecho, existen algunas enzimas que son capaces de llevar a cabo tanto la fosforilación de riboflavina a FMN como la reacción de FMN a FAD.[32] La riboflavina cinasa puede ayudar a prevenir un accidente cerebrovascular y posiblemente podría usarse como tratamiento en el futuro.[33] También está implicado en la infección, cuando se estudia en ratones.[34]
Timidina cinasa
[editar]La timidina cinasa es una de las muchas nucleósido cinasas que son responsables de la fosforilación de nucleósidos. Fosforila la timidina para crear monofosfato de timidina (dTMP). Esta cinasa usa una molécula de ATP para suministrar el fosfato a la timidina, como se muestra a continuación. Esta transferencia de un fosfato de un nucleótido a otro por timidina cinasa, así como otras nucleósidos y nucleótidos cinasas, funciona para ayudar a controlar el nivel de cada uno de los diferentes nucleótidos.

Después de la creación de la molécula dTMP, otra cinasa, timidilato cinasa, puede actuar sobre dTMP para crear la forma difosfato, dTDP. El nucleósido difosfato cinasa cataliza la producción de timidina trifosfato, dTTP, que se utiliza en la síntesis de ADN. Debido a esto, la actividad de la timidina cinasa está estrechamente relacionada con el ciclo celular y se usa como marcador tumoral en la química clínica.[35] Por lo tanto, en ocasiones se puede utilizar para predecir el pronóstico del paciente.[36] Los pacientes con mutaciones en el gen de la timidina cinasa pueden tener cierto tipo de síndrome de depleción del ADN mitocondrial, una enfermedad que conduce a la muerte en la primera infancia.[37]
Véase también
[editar]- Bucle de activación
- Señal telefónica
- cinasa dependiente de ciclina
- Receptor acoplado a proteína G
- Fosfatasa
- Fosfolípido
- Fosfoproteína
- Fosforilación
- Fosfotransferasa
- Transducción de señales
- Timidina cinasa
Referencias
[editar]- ↑ Siebold, C; Arnold, I; Garcia-Alles, LF; Baumann, U; Erni, B (28 de noviembre de 2003). «Crystal structure of the Citrobacter freundii dihydroxyacetone kinase reveals an eight-stranded alpha-helical barrel ATP-binding domain.». The Journal of Biological Chemistry 278 (48): 48236-44. PMID 12966101. doi:10.1074/jbc.M305942200.
- ↑ a b Real Academia Nacional de Medicina. «Cinasa». dtme.ranm.es. Consultado el 25 de julio de 2021.
- ↑ a b «The protein kinase complement of the human genome». Science 298 (5600): 1912-1934. 2002. PMID 12471243. doi:10.1126/science.1075762.
- ↑ «Kinase». TheFreeDictionary.com
- ↑ «History of ATP research milestones from an ATP-related chemistry». Nobelprize.org.
- ↑ «Day of the dead: pseudokinases and pseudophosphatases in physiology and disease.». Trends in Cell Biology 24 (9): 489-505. 2014. PMID 24818526. doi:10.1016/j.tcb.2014.03.008.
- ↑ Foulkes DM, Byrne DP and Eyers PA (2017) Pseudokinases: update on their functions and evaluation as new drug targets. Future Med Chem. 9(2):245-265
- ↑ Samarasinghe, Buddhini. «Hallmarks of Cancer 1». Scientific American.
- ↑ Bleeker, FE; Lamba, S; Zanon, C; Molenaar, RJ; Hulsebos, TJ; Troost, D; van Tilborg, AA; Vandertop, WP et al. (26 de septiembre de 2014). «Mutational profiling of kinases in glioblastoma.». BMC Cancer 14: 718. PMC 4192443. PMID 25256166. doi:10.1186/1471-2407-14-718.
- ↑ Lahiry, Piya; Torkamani, Ali; Schork, Nicholas J.; Hegele, Robert A. (January 2010). «Kinase mutations in human disease: interpreting genotype–phenotype relationships». Nature Reviews Genetics 11 (1): 60-74. PMID 20019687. doi:10.1038/nrg2707.
- ↑ Krebs, EG (5 de julio de 1983). «Historical perspectives on protein phosphorylation and a classification system for protein kinases.». Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 302 (1108): 3-11. PMID 6137005. doi:10.1098/rstb.1983.0033.
- ↑ Corbellino, M; Poirel, L; Aubin, JT; Paulli, M; Magrini, U; Bestetti, G; Galli, M; Parravicini, C (Jun 1996). «The role of human herpesvirus 8 and Epstein-Barr virus in the pathogenesis of giant lymph node hyperplasia (Castleman's disease).». Clinical Infectious Diseases 22 (6): 1120-1. PMID 8783733. doi:10.1093/clinids/22.6.1120.
- ↑ Scheeff, Eric D.; Bourne, Philip E. (2005). «Structural Evolution of the Protein Kinase–Like Superfamily». PLoS Computational Biology 1 (5): e49. PMC 1261164. PMID 16244704. doi:10.1371/journal.pcbi.0010049.
- ↑ a b Krebs, EG; Tan, ST; Carrow, DJ; Watts, MK (Oct 1985). «The phosphorylation of proteins: a major mechanism for biological regulation. Fourteenth Sir Frederick Gowland Hopkins memorial lecture.». Biochemical Society Transactions 13 (5): 813-20. PMID 2998902. doi:10.1042/bst0130813.
- ↑ Manning, G; Whyte, DB; Martinez, R; Hunter, T; Sudarsanam, S (Dec 6, 2002). «The protein kinase complement of the human genome.». Science 298 (5600): 1912-34. PMID 12471243. doi:10.1126/science.1075762.
- ↑ Harper, J. W.; Adams, P. D. (August 2001). «Cyclin-Dependent Kinases». Chemical Reviews 101 (8): 2511-2526. PMID 11749386. doi:10.1021/cr0001030.
- ↑ Karp, Gerald (2010). Cell and molecular biology: concepts and experiments (6th edición). Hoboken, NJ: John Wiley. ISBN 9780470483374.
- ↑ Lim, S.; Kaldis, P. (16 de julio de 2013). «Cdks, cyclins and CKIs: roles beyond cell cycle regulation». Development 140 (15): 3079-3093. PMID 23861057. doi:10.1242/dev.091744.
- ↑ a b
- ↑ a b Garrington, TP; Johnson, GL (Apr 1999). «Organization and regulation of mitogen-activated protein kinase signaling pathways.». Current Opinion in Cell Biology 11 (2): 211-8. PMID 10209154. doi:10.1016/s0955-0674(99)80028-3.
- ↑ Sun, Yue; Thapa, Narendra; Hedman, Andrew C.; Anderson, Richard A. (June 2013). «Phosphatidylinositol 4,5-bisphosphate: Targeted production and signaling». BioEssays 35 (6): 513-522. PMC 3882169. PMID 23575577. doi:10.1002/bies.201200171.
- ↑ Heath, CM (2003). «Lipid Kinases Play Crucial and Multiple Roles in Membrane Trafficking and Signalling». Histology and Histopathology 18: 989-998.
- ↑ Cantley, Lewis C (2012). «PI 3-kinase and disease». BMC Proceedings 6 (Suppl 3): O2. doi:10.1186/1753-6561-6-S3-O2.
- ↑ a b c Miller, S.; Tavshanjian, B.; Oleksy, A.; Perisic, O.; Houseman, B. T.; Shokat, K. M.; Williams, R. L. (25 de marzo de 2010). «Shaping Development of Autophagy Inhibitors with the Structure of the Lipid Kinase Vps34». Science 327 (5973): 1638-1642. PMC 2860105. PMID 20339072. doi:10.1126/science.1184429.
- ↑ Neubauer, Heidi A.; Pitson, Stuart M. (November 2013). «Roles, regulation and inhibitors of sphingosine kinase 2». FEBS Journal 280 (21): 5317-5336. PMID 23638983. doi:10.1111/febs.12314.
- ↑ Holzer, H; Duntze, W (1971). «Metabolic regulation by chemical modification of enzymes.». Annual Review of Biochemistry 40: 345-74. PMID 4399446. doi:10.1146/annurev.bi.40.070171.002021.
- ↑ «Nonspherocytic hemolytic anemia due to hexokinase deficiency». Archivado desde el original el 5 de septiembre de 2015. Consultado el 25 de julio de 2021.
- ↑ «Phosphofructokinase Deficiency Glycogen Storage Disease». Archivado desde el original el 19 de abril de 2015. Consultado el 25 de julio de 2021.
- ↑ Bauer, S; Kemter, K; Bacher, A; Huber, R; Fischer, M; Steinbacher, S (7 de marzo de 2003). «Crystal structure of Schizosaccharomyces pombe riboflavin kinase reveals a novel ATP and riboflavin-binding fold.». Journal of Molecular Biology 326 (5): 1463-73. PMID 12595258. doi:10.1016/s0022-2836(03)00059-7.
- ↑ Pratt, Donald Voet, Judith G. Voet, Charlotte W. (2008). Fundamentals of biochemistry : life at the molecular level (3rd edición). Hoboken, NJ: Wiley. ISBN 9780470129302.
- ↑ a b Karthikeyan, S; Zhou, Q; Osterman, AL; Zhang, H (4 de noviembre de 2003). «Ligand binding-induced conformational changes in riboflavin kinase: structural basis for the ordered mechanism.». Biochemistry 42 (43): 12532-8. PMID 14580199. doi:10.1021/bi035450t.
- ↑ Galluccio, M; Brizio, C; Torchetti, EM; Ferranti, P; Gianazza, E; Indiveri, C; Barile, M (Mar 2007). «Over-expression in Escherichia coli, purification and characterization of isoform 2 of human FAD synthetase.». Protein Expression and Purification 52 (1): 175-81. PMID 17049878. doi:10.1016/j.pep.2006.09.002.
- ↑ Zou, YX; Zhang, XH; Su, FY; Liu, X (Oct 2012). «Importance of riboflavin kinase in the pathogenesis of stroke.». CNS Neuroscience & Therapeutics 18 (10): 834-40. PMC 6493343. PMID 22925047. doi:10.1111/j.1755-5949.2012.00379.x.
- ↑ Brijlal, Sangeetha; Lakshmi, A. V; Bamji, Mahtab S.; Suresh, P. (9 de marzo de 2007). «Flavin metabolism during respiratory infection in mice». British Journal of Nutrition 76 (3): 453-62. PMID 8881717. doi:10.1079/BJN19960050.
- ↑ Aufderklamm, S; Todenhöfer, T; Gakis, G; Kruck, S; Hennenlotter, J; Stenzl, A; Schwentner, C (Mar 2012). «Thymidine kinase and cancer monitoring.». Cancer Letters 316 (1): 6-10. PMID 22068047. doi:10.1016/j.canlet.2011.10.025.
- ↑ Topolcan, Ondrej; Holubec, Lubos (February 2008). «The role of thymidine kinase in cancer diseases». Expert Opinion on Medical Diagnostics 2 (2): 129-141. PMID 23485133. doi:10.1517/17530059.2.2.129.
- ↑ Gotz, A.; Isohanni, P.; Pihko, H.; Paetau, A.; Herva, R.; Saarenpaa-Heikkila, O.; Valanne, L.; Marjavaara, S. et al. (21 de junio de 2008). «Thymidine kinase 2 defects can cause multi-tissue mtDNA depletion syndrome». Brain 131 (11): 2841-2850. PMID 18819985. doi:10.1093/brain/awn236.