ABC transporter
| ABC Transporter, NBD | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 Vitamin B12 transporter, BtuCD PDB 1l7v | |||||||||
| Identifiers | |||||||||
| Symbol | ABC_tran | ||||||||
| Pfam | PF00005 | ||||||||
| InterPro | IPR003439 | ||||||||
| PROSITE | PDOC00185 | ||||||||
| SCOP2 | 1b0u / SCOPe / SUPFAM | ||||||||
| TCDB | 3.A.1 | ||||||||
| OPM superfamily | 17 | ||||||||
| OPM protein | 3g5u | ||||||||
| |||||||||


The ABC transporters, ATP synthase (ATP)-binding cassette transporters are a transport system superfamily that is one of the largest and possibly one of the oldest gene families. It is represented in all extant phyla, from prokaryotes to humans.[1][2][3] ABC transporters belong to translocases.
ABC transporters often consist of multiple subunits, one or two of which are transmembrane proteins and one or two of which are membrane-associated AAA ATPases.[citation needed] The ATPase subunits utilize the energy of adenosine triphosphate (ATP) binding and hydrolysis to provide the energy needed for the translocation of substrates across membranes, either for uptake or for export of the substrate.
Most of the uptake systems also have an extracytoplasmic receptor, a solute binding protein. Some homologous ATPases function in non-transport-related processes such as translation of RNA and DNA repair.[4][5] ABC transporters are considered to be an ABC superfamily based on the similarities of the sequence and organization of their ATP-binding cassette (ABC) domains, even though the integral membrane proteins appear to have evolved independently several times, and thus comprise different protein families.[6] Like the ABC exporters, it is possible that the integral membrane proteins of ABC uptake systems also evolved at least three times independently, based on their high resolution three-dimensional structures.[7] ABC uptake porters take up a large variety of nutrients, biosynthetic precursors, trace metals and vitamins, while exporters transport lipids, sterols, drugs, and a large variety of primary and secondary metabolites. Some of these exporters in humans are involved in tumor resistance, cystic fibrosis and a range of other inherited human diseases. High level expression of the genes encoding some of these exporters in both prokaryotic and eukaryotic organisms (including human) result in the development of resistance to multiple drugs such as antibiotics and anti-cancer agents.
Hundreds of ABC transporters have been characterized from both prokaryotes and eukaryotes.[8] ABC genes are essential for many processes in the cell, and mutations in human genes cause or contribute to several human genetic diseases.[9] Forty eight ABC genes have been reported in humans. Among these, many have been characterized and shown to be causally related to diseases present in humans such as cystic fibrosis, adrenoleukodystrophy, Stargardt disease, drug-resistant tumors, Dubin–Johnson syndrome, Byler's disease, progressive familiar intrahepatic cholestasis, X-linked sideroblastic anemia, ataxia, and persistent and hyperinsulimenic hypoglycemia.[8] ABC transporters are also involved in multiple drug resistance, and this is how some of them were first identified. When the ABC transport proteins are overexpressed in cancer cells, they can export anticancer drugs and render tumors resistant.[10]
Function
[edit]ABC transporters utilize the energy of ATP binding and hydrolysis to transport various substrates across cellular membranes. They are divided into three main functional categories. In prokaryotes, importers mediate the uptake of nutrients into the cell. The substrates that can be transported include ions, amino acids, peptides, sugars, and other molecules that are mostly hydrophilic. The membrane-spanning region of the ABC transporter protects hydrophilic substrates from the lipids of the membrane bilayer thus providing a pathway across the cell membrane. Eukaryotes do not possess any importers. Exporters or effluxers, which are present both in prokaryotes and eukaryotes, function as pumps that extrude toxins and drugs out of the cell. In gram-negative bacteria, exporters transport lipids and some polysaccharides from the cytoplasm to the periplasm. The third subgroup of ABC proteins do not function as transporters, but are rather involved in translation and DNA repair processes.[4]
Prokaryotic
[edit]Bacterial ABC transporters are essential in cell viability, virulence, and pathogenicity.[1][4] Iron ABC uptake systems, for example, are important effectors of virulence.[11] Pathogens use siderophores, such as Enterobactin, to scavenge iron that is in complex with high-affinity iron-binding proteins or erythrocytes. These are high-affinity iron-chelating molecules that are secreted by bacteria and reabsorb iron into iron-siderophore complexes. The chvE-gguAB gene in Agrobacterium tumefaciens encodes glucose and galactose importers that are also associated with virulence.[12][13] Transporters are extremely vital in cell survival such that they function as protein systems that counteract any undesirable change occurring in the cell. For instance, a potential lethal increase in osmotic strength is counterbalanced by activation of osmosensing ABC transporters that mediate uptake of solutes.[14] Other than functioning in transport, some bacterial ABC proteins are also involved in the regulation of several physiological processes.[4]
In bacterial efflux systems, certain substances that need to be extruded from the cell include surface components of the bacterial cell (e.g. capsular polysaccharides, lipopolysaccharides, and teichoic acid), proteins involved in bacterial pathogenesis (e.g. hemolysis, heme-binding protein, and alkaline protease), heme, hydrolytic enzymes, S-layer proteins, competence factors, toxins, antibiotics, bacteriocins, peptide antibiotics, drugs and siderophores.[15] They also play important roles in biosynthetic pathways, including extracellular polysaccharide biosynthesis[16] and cytochrome biogenesis.[17]
Eukaryotic
[edit]Although most eukaryotic ABC transporters are effluxers, some are not directly involved in transporting substrates. In the cystic fibrosis transmembrane regulator (CFTR) and in the sulfonylurea receptor (SUR), ATP hydrolysis is associated with the regulation of opening and closing of ion channels carried by the ABC protein itself or other proteins.[5]
Human ABC transporters are involved in several diseases that arise from polymorphisms in ABC genes and rarely due to complete loss of function of single ABC proteins.[18] Such diseases include Mendelian diseases and complex genetic disorders such as cystic fibrosis, adrenoleukodystrophy, Stargardt disease, Tangier disease, immune deficiencies, progressive familial intrahepatic cholestasis, Dubin–Johnson syndrome, Pseudoxanthoma elasticum, persistent hyperinsulinemic hypoglycemia of infancy due to focal adenomatous hyperplasia, X-linked sideroblastosis and anemia, age-related macular degeneration, familial hypoapoproteinemia, Retinitis pigmentosum, cone rod dystrophy, and others.[5] The human ABCB (MDR/TAP) family is responsible for multiple drug resistance (MDR) against a variety of structurally unrelated drugs. ABCB1 or MDR1 P-glycoprotein is also involved in other biological processes for which lipid transport is the main function. It is found to mediate the secretion of the steroid aldosterone by the adrenals, and its inhibition blocked the migration of dendritic immune cells,[19] possibly related to the outward transport of the lipid platelet activating factor (PAF). It has also been reported that ABCB1 mediates transport of cortisol and dexamethasone, but not of progesterone in ABCB1 transfected cells. MDR1 can also transport cholesterol, short-chain and long-chain analogs of phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), sphingomyelin (SM), and glucosylceramide (GlcCer). Multispecific transport of diverse endogenous lipids through the MDR1 transporter can possibly affect the transbilayer distribution of lipids, in particular of species normally predominant on the inner plasma membrane leaflet such as PS and PE.[18]
More recently, ABC-transporters have been shown to exist within the placenta, indicating they could play a protective role for the developing fetus against xenobiotics.[20] Evidence has shown that placental expression of the ABC-transporters P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) are increased in preterm compared to term placentae, with P-gp expression further increased in preterm pregnancies with chorioamnionitis.[21] To a lesser extent, increasing maternal BMI also associated with increased placental ABC-transporter expression, but only at preterm.[21]
Structure
[edit]

All ABC transport proteins share a structural organization consisting of four core domains.[22] These domains consist of two trans-membrane (T) domains and two cytosolic (A) domains. The two T domains alternate between an inward and outward facing orientation, and the alternation is powered by the hydrolysis of adenosine triphosphate or ATP. ATP binds to the A subunits and it is then hydrolyzed to power the alternation, but the exact process by which this happens is not known. The four domains can be present in four separate polypeptides, which occur mostly in bacteria, or present in one or two multi-domain polypeptides.[10] When the polypeptides are one domain, they can be referred to as a full domain, and when they are two multi-domains they can be referred to as a half domain.[9] The T domains are each built of typically 10 membrane spanning alpha helices, through which the transported substance can cross through the plasma membrane. Also, the structure of the T domains determines the specificity of each ABC protein. In the inward facing conformation, the binding site on the A domain is open directly to the surrounding aqueous solutions. This allows hydrophilic molecules to enter the binding site directly from the inner leaflet of the phospholipid bilayer. In addition, a gap in the protein is accessible directly from the hydrophobic core of the inner leaflet of the membrane bilayer. This allows hydrophobic molecules to enter the binding site directly from the inner leaflet of the phospholipid bilayer. After the ATP powered move to the outward facing conformation, molecules are released from the binding site and allowed to escape into the exoplasmic leaflet or directly into the extracellular medium.[10]
The common feature of all ABC transporters is that they consist of two distinct domains, the transmembrane domain (TMD) and the nucleotide-binding domain (NBD). The TMD, also known as membrane-spanning domain (MSD) or integral membrane (IM) domain, consists of alpha helices, embedded in the membrane bilayer. It recognizes a variety of substrates and undergoes conformational changes to transport the substrate across the membrane. The sequence and architecture of TMDs is variable, reflecting the chemical diversity of substrates that can be translocated. The NBD or ATP-binding cassette (ABC) domain, on the other hand, is located in the cytoplasm and has a highly conserved sequence. The NBD is the site for ATP binding.[23] In most exporters, the N-terminal transmembrane domain and the C-terminal ABC domains are fused as a single polypeptide chain, arranged as TMD-NBD-TMD-NBD. An example is the E. coli hemolysin exporter HlyB. Importers have an inverted organization, that is, NBD-TMD-NBD-TMD, where the ABC domain is N-terminal whereas the TMD is C-terminal, such as in the E. coli MacB protein responsible for macrolide resistance.[4][5]
The structural architecture of ABC transporters consists minimally of two TMDs and two NBDs. Four individual polypeptide chains including two TMD and two NBD subunits, may combine to form a full transporter such as in the E. coli BtuCD[24][25] importer involved in the uptake of vitamin B12. Most exporters, such as in the multidrug exporter Sav1866[26] from Staphylococcus aureus, are made up of a homodimer consisting of two half transporters or monomers of a TMD fused to a nucleotide-binding domain (NBD). A full transporter is often required to gain functionality. Some ABC transporters have additional elements that contribute to the regulatory function of this class of proteins. In particular, importers have a high-affinity binding protein (BP) that specifically associates with the substrate in the periplasm for delivery to the appropriate ABC transporter. Exporters do not have the binding protein but have an intracellular domain (ICD) that joins the membrane-spanning helices and the ABC domain. The ICD is believed to be responsible for communication between the TMD and NBD.[23]
Transmembrane domain (TMD)
[edit]Most transporters have transmembrane domains that consist of a total of 12 α-helices with 6 α-helices per monomer. Since TMDs are structurally diverse, some transporters have varying number of helices (between six and eleven). The TM domains are categorized into three distinct sets of folds: type I ABC importer, type II ABC importer and ABC exporter folds. The classification of importer folds is based on detailed characterization of the sequences.[23]
The type I ABC importer fold was originally observed in the ModB TM subunit of the molybdate transporter.[27] This diagnostic fold can also be found in the MalF and MalG TM subunits of MalFGK2[28] and the Met transporter MetI.[29] In the MetI transporter, a minimal set of 5 transmembrane helices constitute this fold while an additional helix is present for both ModB and MalG. The common organization of the fold is the "up-down" topology of the TM2-5 helices that lines the translocation pathway and the TM1 helix wrapped around the outer, membrane-facing surface and contacts the other TM helices.
The type II ABC importer fold is observed in the twenty TM helix-domain of BtuCD[24] and in Hi1471,[30] a homologous transporter from Haemophilus influenzae. In BtuCD, the packing of the helices is complex. The noticeable pattern is that the TM2 helix is positioned through the center of the subunit where it is surrounded in close proximity by the other helices. Meanwhile, the TM5 and TM10 helices are positioned in the TMD interface. The membrane spanning region of ABC exporters is organized into two "wings" that are composed of helices TM1 and TM2 from one subunit and TM3-6 of the other, in a domain-swapped arrangement. A prominent pattern is that helices TM1-3 are related to TM4-6 by an approximate twofold rotation around an axis in the plane of the membrane.[23]
The exporter fold is originally observed in the Sav1866 structure. It contains 12 TM helices, 6 per monomer.[23]
Nucleotide-binding domain (NBD)
[edit]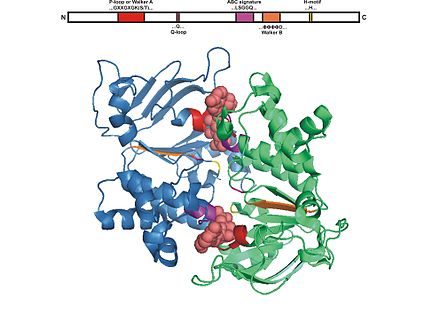
The ABC domain consists of two domains, the catalytic core domain similar to RecA-like motor ATPases and a smaller, structurally diverse α-helical subdomain that is unique to ABC transporters. The larger domain typically consists of two β-sheets and six α helices, where the catalytic Walker A motif (GXXGXGKS/T where X is any amino acid) or P-loop and Walker B motif (ΦΦΦΦD, of which Φ is a hydrophobic residue) is situated. The helical domain consists of three or four helices and the ABC signature motif, also known as LSGGQ motif, linker peptide or C motif. The ABC domain also has a glutamine residue residing in a flexible loop called Q loop, lid or γ-phosphate switch, that connects the TMD and ABC. The Q loop is presumed to be involved in the interaction of the NBD and TMD, particularly in the coupling of nucleotide hydrolysis to the conformational changes of the TMD during substrate translocation. The H motif or switch region contains a highly conserved histidine residue that is also important in the interaction of the ABC domain with ATP. The name ATP-binding cassette is derived from the diagnostic arrangement of the folds or motifs of this class of proteins upon formation of the ATP sandwich and ATP hydrolysis.[4][15][23]
ATP binding and hydrolysis
[edit]Dimer formation of the two ABC domains of transporters requires ATP binding.[31] It is generally observed that the ATP bound state is associated with the most extensive interface between ABC domains, whereas the structures of nucleotide-free transporters exhibit conformations with greater separations between the ABC domains.[23] Structures of the ATP-bound state of isolated NBDs have been reported for importers including HisP,[32] GlcV,[33] MJ1267,[34] E. coli MalK (E.c.MalK),[35] T. litoralis MalK (TlMalK),[36] and exporters such as TAP,[37] HlyB,[38] MJ0796,[39][40] Sav1866,[26] and MsbA.[41] In these transporters, ATP is bound to the ABC domain. Two molecules of ATP are positioned at the interface of the dimer, sandwiched between the Walker A motif of one subunit and the LSGGQ motif of the other.[23] This was first observed in Rad50[42] and reported in structures of MJ0796, the NBD subunit of the LolD transporter from Methanococcus jannaschii[40] and E.c.MalK of a maltose transporter.[35] These structures were also consistent with results from biochemical studies revealing that ATP is in close contact with residues in the P-loop and LSGGQ motif during catalysis.[43]
Nucleotide binding is required to ensure the electrostatic and/or structural integrity of the active site and contribute to the formation of an active NBD dimer.[44] Binding of ATP is stabilized by the following interactions: (1) ring-stacking interaction of a conserved aromatic residue preceding the Walker A motif and the adenosine ring of ATP,[45][46] (2) hydrogen-bonds between a conserved lysine residue in the Walker A motif and the oxygen atoms of the β- and γ-phosphates of ATP and coordination of these phosphates and some residues in the Walker A motif with Mg2+ ion,[33][37] and (3) γ-phosphate coordination with side chain of serine and backbone amide groups of glycine residues in the LSGGQ motif.[47] In addition, a residue that suggests the tight coupling of ATP binding and dimerization, is the conserved histidine in the H-loop. This histidine contacts residues across the dimer interface in the Walker A motif and the D loop, a conserved sequence following the Walker B motif.[35][40][42][48]
The enzymatic hydrolysis of ATP requires proper binding of the phosphates and positioning of the γ-phosphate to the attacking water.[23] In the nucleotide binding site, the oxygen atoms of the β- and γ-phosphates of ATP are stabilized by residues in the Walker A motif[49][50] and coordinate with Mg2+.[23] This Mg2+ ion also coordinates with the terminal aspartate residue in the Walker B motif through the attacking H2O.[33][34][39] A general base, which may be the glutamate residue adjacent to the Walker B motif,[31][40][46] glutamine in the Q-loop,[30][36][40] or a histidine in the switch region that forms a hydrogen bond with the γ-phosphate of ATP, is found to catalyze the rate of ATP hydrolysis by promoting the attacking H2O.[35][36][40][48] The precise molecular mechanism of ATP hydrolysis is still controversial.[4]
Mechanism of transport
[edit]ABC transporters are active transporters, that is, they use energy in the form of adenosine triphosphate (ATP) to translocate substrates across cell membranes. These proteins harness the energy of ATP binding and/or hydrolysis to drive conformational changes in the transmembrane domain (TMD) and consequently transport molecules.[51] ABC importers and exporters have a common mechanism for transporting substrates. They are similar in their structures. The model that describes the conformational changes associated with the binding of the substrate is the alternating-access model. In this model, the substrate binding site alternates between outward- and inward-facing conformations. The relative binding affinities of the two conformations for the substrate largely determines the net direction of transport. For importers, since translocation is directed from the periplasm to the cytoplasm, the outward-facing conformation has higher binding affinity for the substrate. In contrast, the substrate binding affinity in exporters is greater in the inward-facing conformation.[23] A model that describes the conformational changes in the nucleotide-binding domain (NBD) as a result of ATP binding and hydrolysis is the ATP-switch model. This model presents two principal conformations of the NBDs: formation of a closed dimer upon binding two ATP molecules and dissociation to an open dimer facilitated by ATP hydrolysis and release of inorganic phosphate (Pi) and adenosine diphosphate (ADP). Switching between the open and closed dimer conformations induces conformational changes in the TMD resulting in substrate translocation.[52]
The general mechanism for the transport cycle of ABC transporters has not been fully elucidated, but substantial structural and biochemical data has accumulated to support a model in which ATP binding and hydrolysis is coupled to conformational changes in the transporter. The resting state of all ABC transporters has the NBDs in an open dimer configuration, with low affinity for ATP. This open conformation possesses a chamber accessible to the interior of the transporter. The transport cycle is initiated by binding of substrate to the high-affinity site on the TMDs, which induces conformational changes in the NBDs and enhances the binding of ATP. Two molecules of ATP bind, cooperatively, to form the closed dimer configuration. The closed NBD dimer induces a conformational change in the TMDs such that the TMD opens, forming a chamber with an opening opposite to that of the initial state. The affinity of the substrate to the TMD is reduced, thereby releasing the substrate. Hydrolysis of ATP follows and then sequential release of Pi and then ADP restores the transporter to its basal configuration. Although a common mechanism has been suggested, the order of substrate binding, nucleotide binding and hydrolysis, and conformational changes, as well as interactions between the domains is still debated.[4][15][18][23][41][44][51][52][53][54][55]
Several groups studying ABC transporters have differing assumptions on the driving force of transporter function. It is generally assumed that ATP hydrolysis provides the principal energy input or "power stroke" for transport and that the NBDs operate alternately and are possibly involved in different steps in the transport cycle.[56] However, recent structural and biochemical data shows that ATP binding, rather than ATP hydrolysis, provides the "power stroke".[57] It may also be that since ATP binding triggers NBD dimerization, the formation of the dimer may represent the "power stroke." In addition, some transporters have NBDs that do not have similar abilities in binding and hydrolyzing ATP and that the interface of the NBD dimer consists of two ATP binding pockets suggests a concurrent function of the two NBDs in the transport cycle.[52]
Some evidence to show that ATP binding is indeed the power stroke of the transport cycle was reported.[52] It has been shown that ATP binding induces changes in the substrate-binding properties of the TMDs. The affinity of ABC transporters for substrates has been difficult to measure directly, and indirect measurements, for instance through stimulation of ATPase activity, often reflects other rate-limiting steps. Recently, direct measurement of vinblastine binding to permease-glycoprotein (P-glycoprotein) in the presence of nonhydrolyzable ATP analogs, e.g. 5'-adenylyl-β-γ-imidodiphosphate (AMP-PNP), showed that ATP binding, in the absence of hydrolysis, is sufficient to reduce substrate-binding affinity.[58] Also, ATP binding induces substantial conformational changes in the TMDs. Spectroscopic, protease accessibility and crosslinking studies have shown that ATP binding to the NBDs induces conformational changes in multidrug resistance-associated protein-1 (MRP1),[59] HisPMQ,[60] LmrA,[61] and Pgp.[62] Two dimensional crystal structures of AMP-PNP-bound Pgp showed that the major conformational change during the transport cycle occurs upon ATP binding and that subsequent ATP hydrolysis introduces more limited changes.[63] Rotation and tilting of transmembrane α-helices may both contribute to these conformational changes. Other studies have focused on confirming that ATP binding induces NBD closed dimer formation. Biochemical studies of intact transport complexes suggest that the conformational changes in the NBDs are relatively small. In the absence of ATP, the NBDs may be relatively flexible, but they do not involve a major reorientation of the NBDs with respect to the other domains. ATP binding induces a rigid body rotation of the two ABC subdomains with respect to each other, which allows the proper alignment of the nucleotide in the active site and interaction with the designated motifs. There is strong biochemical evidence that binding of two ATP molecules can be cooperative, that is, ATP must bind to the two active site pockets before the NBDs can dimerize and form the closed, catalytically active conformation.[52]
ABC importers
[edit]Most ABC transporters that mediate the uptake of nutrients and other molecules in bacteria rely on a high-affinity solute binding protein (BP). BPs are soluble proteins located in the periplasmic space between the inner and outer membranes of gram-negative bacteria. Gram-positive microorganisms lack a periplasm such that their binding protein is often a lipoprotein bound to the external face of the cell membrane. Some gram-positive bacteria have BPs fused to the transmembrane domain of the transporter itself.[4] The first successful x-ray crystal structure of an intact ABC importer is the molybdenum transporter (ModBC-A) from Archaeoglobus fulgidus.[27] Atomic-resolution structures of three other bacterial importers, E. coli BtuCD,[24] E. coli maltose transporter (MalFGK2-E),[28] and the putative metal-chelate transporter of Haemophilus influenzae, HI1470/1,[30] have also been determined. The structures provided detailed pictures of the interaction of the transmembrane and ABC domains as well as revealed two different conformations with an opening in two opposite directions. Another common feature of importers is that each NBD is bound to one TMD primarily through a short cytoplasmic helix of the TMD, the "coupling helix". This portion of the EAA loop docks in a surface cleft formed between the RecA-like and helical ABC subdomains and lies approximately parallel to the membrane bilayer.[54]
Large ABC importers
[edit]The BtuCD and HI1470/1 are classified as large (Type II) ABC importers. The transmembrane subunit of the vitamin B12 importer, BtuCD, contains 10 TM helices and the functional unit consists of two copies each of the nucleotide binding domain (NBD) and transmembrane domain (TMD). The TMD and NBD interact with one another via the cytoplasmic loop between two TM helices and the Q loop in the ABC. In the absence of nucleotide, the two ABC domains are folded and the dimer interface is open. A comparison of the structures with (BtuCDF) and without (BtuCD) binding protein reveals that BtuCD has an opening that faces the periplasm whereas in BtuCDF, the outward-facing conformation is closed to both sides of the membrane. The structures of BtuCD and the BtuCD homolog, HI1470/1, represent two different conformational states of an ABC transporter. The predicted translocation pathway in BtuCD is open to the periplasm and closed at the cytoplasmic side of the membrane while that of HI1470/1 faces the opposite direction and open only to the cytoplasm. The difference in the structures is a 9° twist of one TM subunit relative to the other.[4][23][54]
Small ABC importers
[edit]Structures of the ModBC-A and MalFGK2-E, which are in complex with their binding protein, correspond to small (Type I) ABC importers. The TMDs of ModBC-A and MalFGK2-E have only six helices per subunit. The homodimer of ModBC-A is in a conformation in which the TM subunits (ModB) orient in an inverted V-shape with a cavity accessible to the cytoplasm. The ABC subunits (ModC), on the other hand, are arranged in an open, nucleotide-free conformation, in which the P-loop of one subunit faces but is detached from the LSGGQ motif of the other. The binding protein ModA is in a closed conformation with substrate bound in a cleft between its two lobes and attached to the extracellular loops of ModB, wherein the substrate is sitting directly above the closed entrance of the transporter. The MalFGK2-E structure resembles the catalytic transition state for ATP hydrolysis. It is in a closed conformation where it contains two ATP molecules, sandwiched between the Walker A and B motifs of one subunit and the LSGGQ motif of the other subunit. The maltose binding protein (MBP or MalE) is docked on the periplasmic side of the TM subunits (MalF and MalG) and a large, occluded cavity can be found at the interface of MalF and MalG. The arrangement of the TM helices is in a conformation that is closed toward the cytoplasm but with an opening that faces outward. The structure suggests a possibility that MBP may stimulate the ATPase activity of the transporter upon binding.[4][23][54]
Mechanism of transport for importers
[edit]
The mechanism of transport for importers supports the alternating-access model. The resting state of importers is inward-facing, where the nucleotide binding domain (NBD) dimer interface is held open by the TMDs and facing outward but occluded from the cytoplasm. Upon docking of the closed, substrate-loaded binding protein towards the periplasmic side of the transmembrane domains, ATP binds and the NBD dimer closes. This switches the resting state of transporter into an outward-facing conformation, in which the TMDs have reoriented to receive substrate from the binding protein. After hydrolysis of ATP, the NBD dimer opens and substrate is released into the cytoplasm. Release of ADP and Pi reverts the transporter into its resting state. The only inconsistency of this mechanism to the ATP-switch model is that the conformation in its resting, nucleotide-free state is different from the expected outward-facing conformation. Although that is the case, the key point is that the NBD does not dimerize unless ATP and binding protein is bound to the transporter.[4][15][23][52][54]
ABC exporters
[edit]Prokaryotic ABC exporters are abundant and have close homologues in eukaryotes. This class of transporters is studied based on the type of substrate that is transported. One class is involved in the protein (e.g. toxins, hydrolytic enzymes, S-layer proteins, lantibiotics, bacteriocins, and competence factors) export and the other in drug efflux. ABC transporters have gained extensive attention because they contribute to the resistance of cells to antibiotics and anticancer agents by pumping drugs out of the cells.[1][64][4] A common mechanism is the overexpression of ABC exporters like P-glycoprotein (P-gp/ABCB1), multidrug resistance-associated protein 1 (MRP1/ABCC1), and breast cancer resistance protein (BCRP/ABCG2) in cancer cells that limit the exposure to anticancer drugs.[65]
In gram-negative organisms, ABC transporters mediate secretion of protein substrates across inner and outer membranes simultaneously without passing through the periplasm. This type of secretion is referred to as type I secretion, which involves three components that function in concert: an ABC exporter, a membrane fusion protein (MFP), and an outer membrane factor (OMF). An example is the secretion of hemolysin (HlyA) from E. coli where the inner membrane ABC transporter HlyB interacts with an inner membrane fusion protein HlyD and an outer membrane facilitator TolC. TolC allows hemolysin to be transported across the two membranes, bypassing the periplasm.[1][64][15]
Bacterial drug resistance has become an increasingly major health problem. One of the mechanisms for drug resistance is associated with an increase in antibiotic efflux from the bacterial cell. Drug resistance associated with drug efflux, mediated by P-glycoprotein, was originally reported in mammalian cells. In bacteria, Levy and colleagues presented the first evidence that antibiotic resistance was caused by active efflux of a drug.[66] P-glycoprotein is the best-studied efflux pump and as such has offered important insights into the mechanism of bacterial pumps.[4] Although some exporters transport a specific type of substrate, most transporters extrude a diverse class of drugs with varying structure.[18] These transporters are commonly called multi-drug resistant (MDR) ABC transporters and sometimes referred to as "hydrophobic vacuum cleaners".[55]
Human ABCB1/MDR1 P-glycoprotein
[edit]P-glycoprotein (3.A.1.201.1) is a well-studied protein associated with multi-drug resistance. It belongs to the human ABCB (MDR/TAP) family and is also known as ABCB1 or MDR1 Pgp. MDR1 consists of a functional monomer with two transmembrane domains (TMD) and two nucleotide-binding domains (NBD). This protein can transport mainly cationic or electrically neutral substrates as well as a broad spectrum of amphiphilic substrates. The structure of the full-size ABCB1 monomer was obtained in the presence and absence of nucleotide using electron cryo crystallography. Without the nucleotide, the TMDs are approximately parallel and form a barrel surrounding a central pore, with the opening facing towards the extracellular side of the membrane and closed at the intracellular face. In the presence of the nonhydrolyzable ATP analog, AMP-PNP, the TMDs have a substantial reorganization with three clearly segregated domains. A central pore, which is enclosed between the TMDs, is slightly open towards the intracellular face with a gap between two domains allowing access of substrate from the lipid phase. Substantial repacking and possible rotation of the TM helices upon nucleotide binding suggests a helix rotation model for the transport mechanism.[18]
Plant transporters
[edit]The genome of the model plant Arabidopsis thaliana is capable of encoding 120 ABC proteins compared to 50-70 ABC proteins that are encoded by the human genome and fruit flies (Drosophila melanogaster). Plant ABC proteins are categorized in 13 subfamilies on the basis of size (full, half or quarter), orientation, and overall amino acid sequence similarity.[67] Multidrug resistant (MDR) homologs, also known as P-glycoproteins, represent the largest subfamily in plants with 22 members and the second largest overall ABC subfamily. The B subfamily of plant ABC transporters (ABCBs) are characterized by their localization to the plasma membrane.[68] Plant ABCB transporters are characterized by heterologously expressing them in Escherichia coli, Saccharomyces cerevisiae, Schizosaccharomyces pombe (fission yeast), and HeLa cells to determine substrate specificity. Plant ABCB transporters have shown to transport the phytohormone indole-3-acetic acid ( IAA),[69] also known as auxin, the essential regulator for plant growth and development.[70][71] The directional polar transport of auxin mediates plant environmental responses through processes such as phototropism and gravitropism.[72] Two of the best studied auxin transporters, ABCB1 and ABCB19, have been characterized to be primary auxin exporters[70] Other ABCB transporters such as ABCB4 participate in both the export and import of auxin[70] At low intracellular auxin concentrations ABCB4 imports auxin until it reaches a certain threshold which then reverses function to only export auxin.[70][73]
Sav1866
[edit]The first high-resolution structure reported for an ABC exporter was that of Sav1866 (3.A.1.106.2) from Staphylococcus aureus.[18][74] Sav1866 is a homolog of multidrug ABC transporters. It shows significant sequence similarity to human ABC transporters of subfamily B that includes MDR1 and TAP1/TAP2. The ATPase activity of Sav1866 is known to be stimulated by cancer drugs such as doxorubicin, vinblastine and others,[75] which suggests similar substrate specificity to P-glycoprotein and therefore a possible common mechanism of substrate translocation. Sav1866 is a homodimer of half transporters, and each subunit contains an N-terminal TMD with six helices and a C-terminal NBD. The NBDs are similar in structure to those of other ABC transporters, in which the two ATP binding sites are formed at the dimer interface between the Walker A motif of one NBD and the LSGGQ motif of the other. The ADP-bound structure of Sav1866 shows the NBDs in a closed dimer and the TM helices split into two "wings" oriented towards the periplasm, forming the outward-facing conformation. Each wing consists of helices TM1-2 from one subunit and TM3-6 from the other subunit. It contains long intracellular loops (ICLs or ICD) connecting the TMDs that extend beyond the lipid bilayer into the cytoplasm and interacts with the 8=D. Whereas the importers contain a short coupling helix that contact a single NBD, Sav1866 has two intracellular coupling helices, one (ICL1) contacting the NBDs of both subunits and the other (ICL2) interacting with only the opposite NBD subunit.[23][26][54]
MsbA
[edit]MsbA (3.A.1.106.1) is a multi-drug resistant (MDR) ABC transporter and possibly a lipid flippase. It is an ATPase that transports lipid A, the hydrophobic moiety of lipopolysaccharide (LPS), a glucosamine-based saccharolipid that makes up the outer monolayer of the outer membranes of most gram-negative bacteria. Lipid A is an endotoxin and so loss of MsbA from the cell membrane or mutations that disrupt transport results in the accumulation of lipid A in the inner cell membrane resulting to cell death. It is a close bacterial homolog of P-glycoprotein (Pgp) by protein sequence homology and has overlapping substrate specificities with the MDR-ABC transporter LmrA from Lactococcus lactis.[76] MsbA from E. coli is 36% identical to the NH2-terminal half of human MDR1, suggesting a common mechanism for transport of amphiphatic and hydrophobic substrates. The MsbA gene encodes a half transporter that contains a transmembrane domain (TMD) fused with a nucleotide-binding domain (NBD). It is assembled as a homodimer with a total molecular mass of 129.2 kD. MsbA contains 6 TMDs on the periplasmic side, an NBD located on the cytoplasmic side of the cell membrane, and an intracellular domain (ICD), bridging the TMD and NBD. This conserved helix extending from the TMD segments into or near the active site of the NBD is largely responsible for crosstalk between TMD and NBD. In particular, ICD1 serves as a conserved pivot about which the NBD can rotate, therefore allowing the NBD to disassociate and dimerize during ATP binding and hydrolysis.[4][15][18][23][44][54][55][77]

Previously published (and now retracted) X-ray structures of MsbA were inconsistent with the bacterial homolog Sav1866.[78][79] The structures were reexamined and found to have an error in the assignment of the hand resulting to incorrect models of MsbA. Recently, the errors have been rectified and new structures have been reported.[41] The resting state of E. coli MsbA exhibits an inverted "V" shape with a chamber accessible to the interior of the transporter suggesting an open, inward-facing conformation. The dimer contacts are concentrated between the extracellular loops and while the NBDs are ≈50Å apart, the subunits are facing each other. The distance between the residues in the site of the dimer interface have been verified by cross-linking experiments[80] and EPR spectroscopy studies.[81] The relatively large chamber allows it to accommodate large head groups such as that present in lipid A. Significant conformational changes are required to move the large sugar head groups across the membrane. The difference between the two nucleotide-free (apo) structures is the ≈30° pivot of TM4/TM5 helices relative to the TM3/TM6 helices. In the closed apo state (from V. cholerae MsbA), the NBDs are aligned and although closer, have not formed an ATP sandwich, and the P loops of opposing monomers are positioned next to one another. In comparison to the open conformation, the dimer interface of the TMDs in the closed, inward-facing conformation has extensive contacts. For both apo conformations of MsbA, the chamber opening is facing inward. The structure of MsbA-AMP-PNP (5'-adenylyl-β-γ-imidodiphosphate), obtained from S. typhimurium, is similar to Sav1866. The NBDs in this nucleotide-bound, outward-facing conformation, come together to form a canonical ATP dimer sandwich, that is, the nucleotide is situated in between the P-loop and LSGGQ motif. The conformational transition from MsbA-closed-apo to MsbA-AMP-PNP involves two steps, which are more likely concerted: a ≈10° pivot of TM4/TM5 helices towards TM3/TM6, bringing the NBDs closer but not into alignment followed by tilting of TM4/TM5 helices ≈20° out of plane. The twisting motion results in the separation of TM3/TM6 helices away from TM1/TM2 leading to a change from an inward- to an outward- facing conformation. Thus, changes in both the orientation and spacing of the NBDs dramatically rearrange the packing of transmembrane helices and effectively switch access to the chamber from the inner to the outer leaflet of the membrane.[41] The structures determined for MsbA is basis for the tilting model of transport.[18] The structures described also highlight the dynamic nature of ABC exporters as also suggested by fluorescence and EPR studies.[54][81][82] Recent work has resulted in the discovery of MsbA inhibitors.[83][84]
Mechanism of transport for exporters
[edit]
ABC exporters have a transport mechanism that is consistent with both the alternating-access model and ATP-switch model. In the apo states of exporters, the conformation is inward-facing and the TMDs and NBDs are relatively far apart to accommodate amphiphilic or hydrophobic substrates. For MsbA, in particular, the size of the chamber is large enough to accommodate the sugar groups from lipopolysaccharides (LPS). As has been suggested by several groups, binding of substrate initiates the transport cycle. The "power stroke", that is, ATP binding that induces NBD dimerization and formation of the ATP sandwich, drives the conformational changes in the TMDs. In MsbA, the sugar head groups are sequestered within the chamber during the "power stroke". The cavity is lined with charged and polar residues that are likely solvated creating an energetically unfavorable environment for hydrophobic substrates and energetically favorable for polar moieties in amphiphilic compounds or sugar groups from LPS. Since the lipid cannot be stable for a long time in the chamber environment, lipid A and other hydrophobic molecules may "flip" into an energetically more favorable position within the outer membrane leaflet. The "flipping" may also be driven by the rigid-body shearing of the TMDs while the hydrophobic tails of the LPS are dragged through the lipid bilayer. Repacking of the helices switches the conformation into an outward-facing state. ATP hydrolysis may widen the periplasmic opening and push the substrate towards the outer leaflet of the lipid bilayer. Hydrolysis of the second ATP molecule and release of Pi separates the NBDs followed by restoration of the resting state, opening the chamber towards the cytoplasm for another cycle.[41][44][52][55][81][85]
Role in multi drug resistance
[edit]ABC transporters are known to play a crucial role in the development of multidrug resistance (MDR). In MDR, patients that are on medication eventually develop resistance not only to the drug they are taking but also to several different types of drugs. This is caused by several factors, one of which is increased expulsion of the drug from the cell by ABC transporters. For example, the ABCB1 protein (P-glycoprotein) functions in pumping tumor suppression drugs out of the cell. Pgp also called MDR1, ABCB1, is the prototype of ABC transporters and also the most extensively-studied gene. Pgp is known to transport organic cationic or neutral compounds. A few ABCC family members, also known as MRP, have also been demonstrated to confer MDR to organic anion compounds. The most-studied member in ABCG family is ABCG2, also known as BCRP (breast cancer resistance protein) confer resistance to most Topoisomerase I or II inhibitors such as topotecan, irinotecan, and doxorubicin.
It is unclear exactly how these proteins can translocate such a wide variety of drugs, however, one model (the hydrophobic vacuum cleaner model) states that, in P-glycoprotein, the drugs are bound indiscriminately from the lipid phase based on their hydrophobicity.
The Discovery of the first eukaryotic ABC transporter protein came from studies on tumor cells and cultured cells that exhibited resistance to several drugs with unrelated chemical structures. These cells were shown to express elevated levels of multidrug-resistance (MDR) transport protein which was originally called P-glycoprotein (P-gp), but it is also referred to as multidrug resistance protein 1 (MDR1) or ABCB1. This protein uses ATP hydrolysis, just like the other ABC transporters, to export a large variety of drugs from the cytosol to the extracellular medium. In multidrug-resistant cells, the MDR1 gene is frequently amplified. This results in a large overproduction of the MDR1 protein. The substrates of mammalian ABCB1 are primarily planar, lipid-soluble molecules with one or more positive charges. All of these substrates compete with one another for transport, suggesting that they bind to the same or overlapping sites on the protein. Many of the drugs that are transported out by ABCB1 are small, nonpolar drugs that diffuse across the extracellular medium into the cytosol, where they block various cellular functions. Drugs such as colchicine and vinblastine, which block assembly of microtubules, freely cross the membrane into the cytosol, but the export of these drugs by ABCB1 reduces their concentration in the cell. Therefore, it takes a higher concentration of the drugs is required to kill the cells that express ABCB1 than those that do not express the gene.[10]
Other ABC transporters that contribute to multidrug resistance are ABCC1 (MRP1) and ABCG2 (breast cancer resistance protein).[86]
To solve the problems associated with multidrug-resistance by MDR1, different types of drugs can be used or the ABC transporters themselves must be inhibited. For other types of drugs to work, they must bypass the resistance mechanism, which is the ABC transporter. To do this other anticancer drugs can be utilized such as alkylating drugs (cyclophosphamide), antimetabolites (5-fluorouracil), and the anthracycline modified drugs (annamycin and doxorubicin-peptide). These drugs would not function as a substrate of ABC transporters, and would thus not be transported. The other option is to use a combination of ABC inhibitory drugs and anticancer drugs at the same time. This would reverse the resistance to the anticancer drugs so that they could function as intended. The substrates that reverse the resistance to anticancer drugs are called chemosensitizers.[8]
Reversal of multi drug resistance
[edit]Drug resistance is a common clinical problem that occurs in patients with infectious diseases and in patients with cancer. Prokaryotic and eukaryotic microorganisms as well as neoplastic cells are often found to be resistant to drugs. MDR is frequently associated with overexpression of ABC transporters. Inhibition of ABC transporters by low-molecular weight compounds has been extensively investigated in cancer patients; however, the clinical results have been disappointing. Recently various RNAi strategies have been applied to reverse MDR in different tumor models and this technology is effective in reversing ABC-transporter-mediated MDR in cancer cells and is therefore a promising strategy for overcoming MDR by gene therapeutic applications. RNAi technology could also be considered for overcoming MDR in infectious diseases caused by microbial pathogens.[87]
Physiological role
[edit]In addition to conferring MDR in tumor cells, ABC transporters are also expressed in the membranes of healthy cells, where they facilitate the transport of various endogenous substances, as well as of substances foreign to the body. For instance, ABC transporters such as Pgp, the MRPs and BCRP limit the absorption of many drugs from the intestine, and pump drugs from the liver cells to the bile[88] as a means of removing foreign substances from the body. A large number of drugs are either transported by ABC transporters themselves or affect the transport of other drugs. The latter scenario can lead to drug-drug interactions,[89] sometimes resulting in altered effects of the drugs.[90]
Methods to characterize ABC transporter interactions
[edit]There are a number of assay types that allow the detection of ABC transporter interactions with endogenous and xenobiotic compounds.[91] The complexity of assay range from relatively simple membrane assays.[92] like vesicular transport assay, ATPase assay to more complex cell based assays up to intricate in vivoJeffrey P, Summerfield SG (2007). "Challenges for blood-brain barrier (BBB) screening". Xenobiotica. 37 (10–11): 1135–51. doi:10.1080/00498250701570285. PMID 17968740. S2CID 25944548. detection methodologies.[93]
Membrane assays
[edit]The vesicular transport assay detects the translocation of molecules by ABC transporters.[94] Membranes prepared under suitable conditions contain inside-out oriented vesicles with the ATP binding site and substrate binding site of the transporter facing the buffer outside. Substrates of the transporter are taken up into the vesicles in an ATP dependent manner. Rapid filtration using glass fiber filters or nitrocellulose membranes are used to separate the vesicles from the incubation solution and the test compound trapped inside the vesicles is retained on the filter. The quantity of the transported unlabelled molecules is determined by HPLC, LC/MS, LC/MS/MS. Alternatively, the compounds are radiolabeled, fluorescent or have a fluorescent tag so that the radioactivity or fluorescence retained on the filter can be quantified.
Various types of membranes from different sources (e.g. insect cells, transfected or selected mammalian cell lines) are used in vesicular transport studies. Membranes are commercially available or can be prepared from various cells or even tissues e.g. liver canalicular membranes. This assay type has the advantage of measuring the actual disposition of the substrate across the cell membrane. Its disadvantage is that compounds with medium-to-high passive permeability are not retained inside the vesicles making direct transport measurements with this class of compounds difficult to perform.
The vesicular transport assay can be performed in an "indirect" setting, where interacting test drugs modulate the transport rate of a reporter compound. This assay type is particularly suitable for the detection of possible drug-drug interactions and drug-endogenous substrate interactions. It is not sensitive to the passive permeability of the compounds and therefore detects all interacting compounds. Yet, it does not provide information on whether the compound tested is an inhibitor of the transporter, or a substrate of the transporter inhibiting its function in a competitive fashion. A typical example of an indirect vesicular transport assay is the detection of the inhibition of taurocholate transport by ABCB11 (BSEP).
Whole cell based assays
[edit]Efflux transporter-expressing cells actively pump substrates out of the cell, which results in a lower rate of substrate accumulation, lower intracellular concentration at steady state, or a faster rate of substrate elimination from cells loaded with the substrate. Transported radioactive substrates or labeled fluorescent dyes can be directly measured, or in an indirect set up, the modulation of the accumulation of a probe substrate (e.g. fluorescent dyes like rhodamine 123, or calcein) can be determined in the presence of a test drug.[89]
Calcein-AM, A highly permeable derivative of calcein readily penetrates into intact cells, where the endogenous esterases rapidly hydrolyze it to the fluorescent calcein. In contrast to calcein-AM, calcein has low permeability and therefore gets trapped in the cell and accumulates. As calcein-AM is an excellent substrate of the MDR1 and MRP1 efflux transporters, cells expressing MDR1 and/or MRP1 transporters pump the calcein-AM out of the cell before esterases can hydrolyze it. This results in a lower cellular accumulation rate of calcein. The higher the MDR activity is in the cell membrane, the less Calcein is accumulated in the cytoplasm. In MDR-expressing cells, the addition of an MDR inhibitor or an MDR substrate in excess dramatically increases the rate of Calcein accumulation. Activity of multidrug transporter is reflected by the difference between the amounts of dye accumulated in the presence and the absence of inhibitor. Using selective inhibitors, transport activity of MDR1 and MRP1 can be easily distinguished. This assay can be used to screen drugs for transporter interactions, and also to quantify the MDR activity of cells. The calcein assay is the proprietary assay of SOLVO Biotechnology.
Subfamilies
[edit]Mammalian subfamilies
[edit]There are 49 known ABC transporters present in humans, which are classified into seven families by the Human Genome Organization.
| Family | Members | Function | Examples |
|---|---|---|---|
| ABCA | This family contains some of the largest transporters (over 2,100 amino acids long). Five of them are located in a cluster in the 17q24 chromosome. | Responsible for the transportation of cholesterol and lipids, among other things. | ABCA12 ABCA1 |
| ABCB | Consists of 4 full and 7 half transporters. | Some are located in the blood–brain barrier, liver, mitochondria, transports peptides and bile, for example. | ABCB5 |
| ABCC | Consists of 12 full transporters. | Used in ion transport, cell-surface receptors, toxin secretion. Includes the CFTR protein, which causes cystic fibrosis when deficient. | ABCC6 |
| ABCD | Consists of 4 half transporters | Are all used in peroxisomes. | ABCD1 |
| ABCE/ABCF | Consists of 1 ABCE and 3 ABCF proteins. | These are not actually transporters but merely ATP-binding domains that were derived from the ABC family, but without the transmembrane domains. These proteins mainly regulate protein synthesis or expression. | ABCE1, ABCF1, ABCF2 |
| ABCG | Consists of 6 "reverse" half-transporters, with the NBF at the NH3+ end and the TM at the COO- end. | Transports lipids, diverse drug substrates, bile, cholesterol, and other steroids. | ABCG2 ABCG1 |
A full list of human ABC transporters can be found from.[95]
ABCA
[edit]The ABCA subfamily is composed of 12 full transporters split into two subgroups. The first subgroup consists of seven genes that map to six different chromosomes. These are ABCA1, ABCA2, ABCA3, and ABCA4, ABCA7, ABCA12, and ABCA13. The other subgroup consists of ABCA5 and ABCA6 and ABCA8, ABCA9 and ABCA10. A8-10. All of subgroup 2 is organized into a head to tail cluster of chromosomes on chromosome 17q24. Genes in this second subgroup are distinguished from ABCA1-like genes by having 37-38 exons as opposed to the 50 exons in ABCA1. The ABCA1 subgroup is implicated in the development of genetic diseases. In the recessive Tangier's disease, the ABCA1 protein is mutated. Also, the ABCA4 maps to a region of chromosome 1p21 that contains the gene for Stargardt's disease. This gene is found to be highly expressed in rod photoreceptors and is mutated in Stargardt's disease, recessive retinitis pigmentism, and the majority of recessive cone-rod dystrophy.[9]
ABCB
[edit]The ABCB subfamily is composed of four full transporters and two half transporters. This is the only human subfamily to have both half and full types of transporters. ABCB1 was discovered as a protein overexpressed in certain drug resistant tumor cells. It is expressed primarily in the blood–brain barrier and liver and is thought to be involved in protecting cells from toxins. Cells that overexpress this protein exhibit multi-drug resistance.[9]
ABCC
[edit]Subfamily ABCC contains thirteen members and nine of these transporters are referred to as the Multidrug Resistance Proteins (MRPs). The MRP proteins are found throughout nature and they mediate many important functions.[96] They are known to be involved in ion transport, toxin secretion, and signal transduction.[9] Of the nine MRP proteins, four of them, MRP4, 5, 8, 9, (ABCC4, 5, 11, and 12), have a typical ABC structure with four domains, comprising two membrane spanning domains, with each spanning domain followed by a nucleotide binding domain. These are referred to as short MRPs. The remaining 5 MRP's (MRP1, 2, 6, 7) (ABCC1, 2, 3, 6 and 10) are known as long MRPs and feature an additional fifth domain at their N terminus.[96]
CFTR, the transporter involved in the disease cystic fibrosis, is also considered part of this subfamily. Cystic fibrosis occurs upon mutation and loss of function of CFTR.[9]
The sulfonylurea receptors (SUR), involved in insulin secretion, neuronal function, and muscle function, are also part of this family of proteins. Mutations in SUR proteins are a potential cause of Neonatal diabetes mellitus. SUR is also the binding site for drugs such as sulfonylureas and potassium-channel openers activators such as diazoxide.
ABCD
[edit]The ABCD subfamily consists of four genes that encode half transporters expressed exclusively in the peroxisome. ABCD1 is responsible for the X-linked form of Adrenoleukodystrophy (ALD) which is a disease characterized by neurodegeneration and adrenal deficiency that typically is initiated in late childhood. The cells of ALD patients feature accumulation of unbranched saturated fatty acids, but the exact role of ABCD1 in the process is still undetermined. In addition, the function of other ABCD genes have yet to be determined but have been thought to exert related functions in fatty acid metabolism.[9]
ABCE and ABCF
[edit]Both of these subgroups are composed of genes that have ATP binding domains that are closely related to other ABC transporters, but these genes do not encode for trans-membrane domains. ABCE consists of only one member, OABP or ABCE1, which is known to recognize certain oligodendrocytes produced in response to certain viral infections. Each member of the ABCF subgroup consist of a pair of ATP binding domains.[9]
ABCG
[edit]Six half transporters with ATP binding sites on the N terminus and trans-membrane domains at the C terminus make up the ABCG subfamily. This orientation is opposite of all other ABC genes. There are only 5 ABCG genes in the human genome, but there are 15 in the Drosophila genome and 10 in yeast. The ABCG2 gene was discovered in cell lines selected for high level resistance for mitoxantrone and no expression of ABCB1 or ABCC1. ABCG2 can export anthracycline anticancer drugs, as well as topotecan, mitoxantrone, or doxorubicin as substrates. Chromosomal translocations have been found to cause the ABCG2 amplification or rearrangement found in resistant cell lines.[9]
Cross-species subfamilies
[edit]This section is missing information about Pfam/InterPro mapping (bit hard to make, need them to improve data too). (December 2020) |
The following classification system for transmembrane solute transporters has been constructed in the TCDB.[97]
Three families of ABC exporters are defined by their evolutionary origins.[6] ABC1 exporters evolved by intragenic triplication of a 2 TMS precursor (TMS = transmembrane segment. A "2 TMS" protein has 2 transmembrane segments) to give 6 TMS proteins. ABC2 exporters evolved by intragenic duplication of a 3 TMS precursor, and ABC3 exporters evolved from a 4 TMS precursor which duplicated either extragenicly to give two 4 TMS proteins, both required for transport function, or intragenicly to give 8 or 10 TMS proteins. The 10 TMS proteins appear to have two extra TMSs between the two 4 TMS repeat units.[98] Most uptake systems (all except 3.A.1.21) are of the ABC2 type, divided into type I and type II by the way they handle nucleotides. A special subfamily of ABC2 importers called ECF use a separate subunit for substrate recognition.[99]
- 3.A.1.106 The Lipid Exporter (LipidE) Family
- 3.A.1.108 The β-Glucan Exporter (GlucanE) Family
- 3.A.1.109 The Protein-1 Exporter (Prot1E) Family
- 3.A.1.110 The Protein-2 Exporter (Prot2E) Family
- 3.A.1.111 The Peptide-1 Exporter (Pep1E) Family
- 3.A.1.112 The Peptide-2 Exporter (Pep2E) Family
- 3.A.1.113 The Peptide-3 Exporter (Pep3E) Family
- 3.A.1.117 The Drug Exporter-2 (DrugE2) Family
- 3.A.1.118 The Microcin J25 Exporter (McjD) Family
- 3.A.1.119 The Drug/Siderophore Exporter-3 (DrugE3) Family
- 3.A.1.123 The Peptide-4 Exporter (Pep4E) Family
- 3.A.1.127 The AmfS Peptide Exporter (AmfS-E) Family
- 3.A.1.129 The CydDC Cysteine Exporter (CydDC-E) Family
- 3.A.1.135 The Drug Exporter-4 (DrugE4) Family
- 3.A.1.139 The UDP-Glucose Exporter (U-GlcE) Family (UPF0014 Family)
- 3.A.1.201 The Multidrug Resistance Exporter (MDR) Family (ABCB)
- 3.A.1.202 The Cystic Fibrosis Transmembrane Conductance Exporter (CFTR) Family (ABCC)
- 3.A.1.203 The Peroxysomal Fatty Acyl CoA Transporter (P-FAT) Family (ABCD)
- 3.A.1.206 The a-Factor Sex Pheromone Exporter (STE) Family (ABCB)
- 3.A.1.208 The Drug Conjugate Transporter (DCT) Family (ABCC) (Dębska et al., 2011)
- 3.A.1.209 The MHC Peptide Transporter (TAP) Family (ABCB)
- 3.A.1.210 The Heavy Metal Transporter (HMT) Family (ABCB)
- 3.A.1.212 The Mitochondrial Peptide Exporter (MPE) Family (ABCB)
- 3.A.1.21 The Siderophore-Fe3+ Uptake Transporter (SIUT) Family
ABC2 (InterPro: IPR000412 [partial]):
- 3.A.1.101 The Capsular Polysaccharide Exporter (CPSE) Family
- 3.A.1.102 The Lipooligosaccharide Exporter (LOSE) Family
- 3.A.1.103 The Lipopolysaccharide Exporter (LPSE) Family
- 3.A.1.104 The Teichoic Acid Exporter (TAE) Family
- 3.A.1.105 The Drug Exporter-1 (DrugE1) Family
- 3.A.1.107 The Putative Heme Exporter (HemeE) Family
- 3.A.1.115 The Na+ Exporter (NatE) Family
- 3.A.1.116 The Microcin B17 Exporter (McbE) Family
- 3.A.1.124 The 3-component Peptide-5 Exporter (Pep5E) Family
- 3.A.1.126 The β-Exotoxin I Exporter (βETE) Family
- 3.A.1.128 The SkfA Peptide Exporter (SkfA-E) Family
- 3.A.1.130 The Multidrug/Hemolysin Exporter (MHE) Family
- 3.A.1.131 The Bacitracin Resistance (Bcr) Family
- 3.A.1.132 The Gliding Motility ABC Transporter (Gld) Family
- 3.A.1.133 The Peptide-6 Exporter (Pep6E) Family
- 3.A.1.138 The Unknown ABC-2-type (ABC2-1) Family
- 3.A.1.141 The Ethyl Viologen Exporter (EVE) Family (DUF990 Family; InterPro: IPR010390)
- 3.A.1.142 The Glycolipid Flippase (G.L.Flippase) Family
- 3.A.1.143 The Exoprotein Secretion System (EcsAB(C))
- 3.A.1.144: Functionally Uncharacterized ABC2-1 (ABC2-1) Family
- 3.A.1.145: Peptidase Fused Functionally Uncharacterized ABC2-2 (ABC2-2) Family
- 3.A.1.146: The actinorhodin (ACT) and undecylprodigiosin (RED) exporter (ARE) family
- 3.A.1.147: Functionally Uncharacterized ABC2-2 (ABC2-2) Family
- 3.A.1.148: Functionally Uncharacterized ABC2-3 (ABC2-3) Family
- 3.A.1.149: Functionally Uncharacterized ABC2-4 (ABC2-4) Family
- 3.A.1.150: Functionally Uncharacterized ABC2-5 (ABC2-5) Family
- 3.A.1.151: Functionally Uncharacterized ABC2-6 (ABC2-6) Family
- 3.A.1.152: The lipopolysaccharide export (LptBFG) Family (InterPro: IPR005495)
- 3.A.1.204 The Eye Pigment Precursor Transporter (EPP) Family (ABCG)
- 3.A.1.205 The Pleiotropic Drug Resistance (PDR) Family (ABCG)
- 3.A.1.211 The Cholesterol/Phospholipid/Retinal (CPR) Flippase Family (ABCA)
- 9.B.74 The Phage Infection Protein (PIP) Family
- all uptake systems (3.A.1.1 - 3.A.1.34 except 3.A.1.21)
- 3.A.1.1 Carbohydrate Uptake Transporter-1 (CUT1)
- 3.A.1.2 Carbohydrate Uptake Transporter-2 (CUT2)
- 3.A.1.3 Polar Amino Acid Uptake Transporter (PAAT)
- 3.A.1.4 Hydrophobic Amino Acid Uptake Transporter (HAAT)
- 3.A.1.5 Peptide/Opine/Nickel Uptake Transporter (PepT)
- 3.A.1.6 Sulfate/Tungstate Uptake Transporter (SulT)
- 3.A.1.7 Phosphate Uptake Transporter (PhoT)
- 3.A.1.8 Molybdate Uptake Transporter (MolT)
- 3.A.1.9 Phosphonate Uptake Transporter (PhnT)
- 3.A.1.10 Ferric Iron Uptake Transporter (FeT)
- 3.A.1.11 Polyamine/Opine/Phosphonate Uptake Transporter (POPT)
- 3.A.1.12 Quaternary Amine Uptake Transporter (QAT)
- 3.A.1.13 Vitamin B12 Uptake Transporter (B12T)
- 3.A.1.14 Iron Chelate Uptake Transporter (FeCT)
- 3.A.1.15 Manganese/Zinc/Iron Chelate Uptake Transporter (MZT)
- 3.A.1.16 Nitrate/Nitrite/Cyanate Uptake Transporter (NitT)
- 3.A.1.17 Taurine Uptake Transporter (TauT)
- 3.A.1.19 Thiamin Uptake Transporter (ThiT)
- 3.A.1.20 Brachyspira Iron Transporter (BIT)
- 3.A.1.21 Siderophore-Fe3+ Uptake Transporter (SIUT)
- 3.A.1.24 The Methionine Uptake Transporter (MUT) Family (Similar to 3.A.1.3 and 3.A.1.12)
- 3.A.1.27 The γ-Hexachlorocyclohexane (HCH) Family (Similar to 3.A.1.24 and 3.A.1.12)
- 3.A.1.34 The Tryptophan (TrpXYZ) Family
- ECF uptake systems
- 3.A.1.18 The Cobalt Uptake Transporter (CoT) Family
- 3.A.1.22 The Nickel Uptake Transporter (NiT) Family
- 3.A.1.23 The Nickel/Cobalt Uptake Transporter (NiCoT) Family
- 3.A.1.25 The Biotin Uptake Transporter (BioMNY) Family
- 3.A.1.26 The Putative Thiamine Uptake Transporter (ThiW) Family
- 3.A.1.28 The Queuosine (Queuosine) Family
- 3.A.1.29 The Methionine Precursor (Met-P) Family
- 3.A.1.30 The Thiamin Precursor (Thi-P) Family
- 3.A.1.31 The Unknown-ABC1 (U-ABC1) Family
- 3.A.1.32 The Cobalamin Precursor (B12-P) Family
- 3.A.1.33 The Methylthioadenosine (MTA) Family
- 3.A.1.114 The Probable Glycolipid Exporter (DevE) Family
- 3.A.1.122 The Macrolide Exporter (MacB) Family
- 3.A.1.125 The Lipoprotein Translocase (LPT) Family
- 3.A.1.134 The Peptide-7 Exporter (Pep7E) Family
- 3.A.1.136 The Uncharacterized ABC-3-type (U-ABC3-1) Family
- 3.A.1.137 The Uncharacterized ABC-3-type (U-ABC3-2) Family
- 3.A.1.140 The FtsX/FtsE Septation (FtsX/FtsE) Family
- 3.A.1.207 The Eukaryotic ABC3 (E-ABC3) Family
Images
[edit]Many structures of water-soluble domains of ABC proteins have been produced in recent years.[2]
See also
[edit]- ATP-binding domain of ABC transporters
- Transmembrane domain of ABC transporters
- Elizabeth P. Carpenter, British structural biologist, first to describe structure of human ABC-transporter ABC10
References
[edit]- ^ a b c d Fath, M. J.; Kolter, R. (December 1993). "ABC transporters: bacterial exporters". Microbiological Reviews. 57 (4): 995–1017. doi:10.1128/MMBR.57.4.995-1017.1993. ISSN 0146-0749. PMC 372944. PMID 8302219.
- ^ a b Jones PM, George AM (Mar 2004). "The ABC transporter structure and mechanism: perspectives on recent research". Cellular and Molecular Life Sciences. 61 (6): 682–99. doi:10.1007/s00018-003-3336-9. PMC 11138499. PMID 15052411. S2CID 21422822.
- ^ Ponte-Sucre A, ed. (2009). ABC Transporters in Microorganisms. Caister Academic. ISBN 978-1-904455-49-3.
- ^ a b c d e f g h i j k l m n o Davidson AL, Dassa E, Orelle C, Chen J (Jun 2008). "Structure, function, and evolution of bacterial ATP-binding cassette systems". Microbiology and Molecular Biology Reviews. 72 (2): 317–64, table of contents. doi:10.1128/MMBR.00031-07. PMC 2415747. PMID 18535149.
- ^ a b c d Goffeau A, de Hertogh B, Baret PV (2013). "ABC Transporters". In Lane WJ, Lennarz MD (eds.). Encyclopedia of Biological Chemistry (Second ed.). London: Academic Press. pp. 7–11. doi:10.1016/B978-0-12-378630-2.00224-3. ISBN 978-0-12-378631-9.
- ^ a b Wang B, Dukarevich M, Sun EI, Yen MR, Saier MH (Sep 2009). "Membrane porters of ATP-binding cassette transport systems are polyphyletic". The Journal of Membrane Biology. 231 (1): 1–10. doi:10.1007/s00232-009-9200-6. PMC 2760711. PMID 19806386.
- ^ ter Beek J, Guskov A, Slotboom DJ (Apr 2014). "Structural diversity of ABC transporters". The Journal of General Physiology. 143 (4): 419–35. doi:10.1085/jgp.201411164. PMC 3971661. PMID 24638992.
- ^ a b c Choi CH (Oct 2005). "ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal". Cancer Cell International. 5: 30. doi:10.1186/1475-2867-5-30. PMC 1277830. PMID 16202168.
- ^ a b c d e f g h i Dean M, Hamon Y, Chimini G (Jul 2001). "The human ATP-binding cassette (ABC) transporter superfamily". Journal of Lipid Research. 42 (7): 1007–17. doi:10.1016/S0022-2275(20)31588-1. PMID 11441126.
- ^ a b c d Scott MP, Lodish HF, Berk A, Kaiser, C, Krieger M, Bretscher A, Ploegh H, Amon A (2012). Molecular Cell Biology. San Francisco: W. H. Freeman. ISBN 978-1-4292-3413-9.
- ^ Henderson DP, Payne SM (Nov 1994). "Vibrio cholerae iron transport systems: roles of heme and siderophore iron transport in virulence and identification of a gene associated with multiple iron transport systems". Infection and Immunity. 62 (11): 5120–5. doi:10.1128/IAI.62.11.5120-5125.1994. PMC 303233. PMID 7927795.
- ^ Cangelosi GA, Ankenbauer RG, Nester EW (Sep 1990). "Sugars induce the Agrobacterium virulence genes through a periplasmic binding protein and a transmembrane signal protein". Proceedings of the National Academy of Sciences of the United States of America. 87 (17): 6708–12. Bibcode:1990PNAS...87.6708C. doi:10.1073/pnas.87.17.6708. PMC 54606. PMID 2118656.
- ^ Kemner JM, Liang X, Nester EW (Apr 1997). "The Agrobacterium tumefaciens virulence gene chvE is part of a putative ABC-type sugar transport operon". Journal of Bacteriology. 179 (7): 2452–8. doi:10.1128/jb.179.7.2452-2458.1997. PMC 178989. PMID 9079938.
- ^ Poolman B, Spitzer JJ, Wood JM (Nov 2004). "Bacterial osmosensing: roles of membrane structure and electrostatics in lipid-protein and protein-protein interactions" (PDF). Biochimica et Biophysica Acta (BBA) - Biomembranes. 1666 (1–2): 88–104. doi:10.1016/j.bbamem.2004.06.013. PMID 15519310. S2CID 21763870.
- ^ a b c d e f Davidson AL, Chen J (2004). "ATP-binding cassette transporters in bacteria". Annual Review of Biochemistry. 73: 241–68. doi:10.1146/annurev.biochem.73.011303.073626. PMID 15189142.
- ^ Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CR (May 1998). "Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis". The Journal of Biological Chemistry. 273 (20): 12466–75. doi:10.1074/jbc.273.20.12466. hdl:2434/611267. PMID 9575204.
- ^ Poole RK, Gibson F, Wu G (Apr 1994). "The cydD gene product, component of a heterodimeric ABC transporter, is required for assembly of periplasmic cytochrome c and of cytochrome bd in Escherichia coli". FEMS Microbiology Letters. 117 (2): 217–23. doi:10.1111/j.1574-6968.1994.tb06768.x. PMID 8181727.
- ^ a b c d e f g h Pohl A, Devaux PF, Herrmann A (Mar 2005). "Function of prokaryotic and eukaryotic ABC proteins in lipid transport". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1733 (1): 29–52. doi:10.1016/j.bbalip.2004.12.007. PMID 15749056.
- ^ Randolph GJ (2001). "Dendritic cell migration to lymph nodes: cytokines, chemokines, and lipid mediators". Seminars in Immunology. 13 (5): 267–74. doi:10.1006/smim.2001.0322. PMID 11502161.
- ^ Gedeon C, Behravan J, Koren G, Piquette-Miller M (2006). "Transport of glyburide by placental ABC transporters: implications in fetal drug exposure". Placenta. 27 (11–12): 1096–102. doi:10.1016/j.placenta.2005.11.012. PMID 16460798.
- ^ a b Scott, Hailey; Martinelli, Lilian M.; Grynspan, David; Bloise, Enrrico; Connor, Kristin L. (2022-03-24). "Preterm Birth Associates With Increased Placental Expression of MDR Transporters Irrespective of Prepregnancy BMI". The Journal of Clinical Endocrinology and Metabolism. 107 (4): 1140–1158. doi:10.1210/clinem/dgab813. ISSN 1945-7197. PMID 34748636. S2CID 243863723.
- ^ Shuman HA (1982). "Active transport of maltose in Escherichia coli K12. Role of the periplasmic maltose-binding protein and evidence for a substrate recognition site in the cytoplasmic membrane". J. Biol. Chem. 257 (10): 5455–61. doi:10.1016/S0021-9258(19)83799-7. PMID 7040366.
- ^ a b c d e f g h i j k l m n o p q Rees DC, Johnson E, Lewinson O (Mar 2009). "ABC transporters: the power to change". Nature Reviews Molecular Cell Biology. 10 (3): 218–27. doi:10.1038/nrm2646. PMC 2830722. PMID 19234479.
- ^ a b c Locher KP, Lee AT, Rees DC (May 2002). "The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism" (PDF). Science. 296 (5570): 1091–8. Bibcode:2002Sci...296.1091L. doi:10.1126/science.1071142. PMID 12004122. S2CID 906489.
- ^ Hvorup RN, Goetz BA, Niederer M, Hollenstein K, Perozo E, Locher KP (Sep 2007). "Asymmetry in the structure of the ABC transporter-binding protein complex BtuCD-BtuF". Science. 317 (5843): 1387–90. Bibcode:2007Sci...317.1387H. doi:10.1126/science.1145950. PMID 17673622. S2CID 37232959.
- ^ a b c Dawson RJ, Locher KP (Sep 2006). "Structure of a bacterial multidrug ABC transporter". Nature. 443 (7108): 180–5. Bibcode:2006Natur.443..180D. doi:10.1038/nature05155. PMID 16943773. S2CID 27132450.
- ^ a b c Hollenstein K, Frei DC, Locher KP (Mar 2007). "Structure of an ABC transporter in complex with its binding protein". Nature. 446 (7132): 213–6. Bibcode:2007Natur.446..213H. doi:10.1038/nature05626. PMID 17322901. S2CID 4417002.
- ^ a b Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J (Nov 2007). "Crystal structure of a catalytic intermediate of the maltose transporter". Nature. 450 (7169): 515–21. Bibcode:2007Natur.450..515O. doi:10.1038/nature06264. PMID 18033289. S2CID 4384771.
- ^ Kadaba NS, Kaiser JT, Johnson E, Lee A, Rees DC (Jul 2008). "The high-affinity E. coli methionine ABC transporter: structure and allosteric regulation". Science. 321 (5886): 250–3. Bibcode:2008Sci...321..250K. doi:10.1126/science.1157987. PMC 2527972. PMID 18621668.
- ^ a b c d Pinkett HW, Lee AT, Lum P, Locher KP, Rees DC (Jan 2007). "An inward-facing conformation of a putative metal-chelate-type ABC transporter" (PDF). Science. 315 (5810): 373–7. doi:10.1126/science.1133488. PMID 17158291. S2CID 10531462.
- ^ a b Moody JE, Millen L, Binns D, Hunt JF, Thomas PJ (Jun 2002). "Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters". The Journal of Biological Chemistry. 277 (24): 21111–4. doi:10.1074/jbc.C200228200. PMC 3516282. PMID 11964392.
- ^ Hung LW, Wang IX, Nikaido K, Liu PQ, Ames GF, Kim SH (Dec 1998). "Crystal structure of the ATP-binding subunit of an ABC transporter". Nature. 396 (6712): 703–7. Bibcode:1998Natur.396..703H. doi:10.1038/25393. PMID 9872322. S2CID 204996524.
- ^ a b c Verdon G, Albers SV, Dijkstra BW, Driessen AJ, Thunnissen AM (Jul 2003). "Crystal structures of the ATPase subunit of the glucose ABC transporter from Sulfolobus solfataricus: nucleotide-free and nucleotide-bound conformations". Journal of Molecular Biology. 330 (2): 343–58. doi:10.1016/S0022-2836(03)00575-8. PMID 12823973.
- ^ a b Karpowich N, Martsinkevich O, Millen L, Yuan YR, Dai PL, MacVey K, Thomas PJ, Hunt JF (Jul 2001). "Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter". Structure. 9 (7): 571–86. doi:10.1016/S0969-2126(01)00617-7. PMID 11470432.
- ^ a b c d Chen J, Lu G, Lin J, Davidson AL, Quiocho FA (Sep 2003). "A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle". Molecular Cell. 12 (3): 651–61. doi:10.1016/j.molcel.2003.08.004. PMID 14527411.
- ^ a b c Diederichs K, Diez J, Greller G, Müller C, Breed J, Schnell C, Vonrhein C, Boos W, Welte W (Nov 2000). "Crystal structure of MalK, the ATPase subunit of the trehalose/maltose ABC transporter of the archaeon Thermococcus litoralis". The EMBO Journal. 19 (22): 5951–61. doi:10.1093/emboj/19.22.5951. PMC 305842. PMID 11080142.
- ^ a b Gaudet R, Wiley DC (Sep 2001). "Structure of the ABC ATPase domain of human TAP1, the transporter associated with antigen processing". The EMBO Journal. 20 (17): 4964–72. doi:10.1093/emboj/20.17.4964. PMC 125601. PMID 11532960.
- ^ Schmitt L, Benabdelhak H, Blight MA, Holland IB, Stubbs MT (Jul 2003). "Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: identification of a variable region within ABC helical domains". Journal of Molecular Biology. 330 (2): 333–42. doi:10.1016/S0022-2836(03)00592-8. PMID 12823972.
- ^ a b Yuan YR, Blecker S, Martsinkevich O, Millen L, Thomas PJ, Hunt JF (Aug 2001). "The crystal structure of the MJ0796 ATP-binding cassette. Implications for the structural consequences of ATP hydrolysis in the active site of an ABC transporter". The Journal of Biological Chemistry. 276 (34): 32313–21. doi:10.1074/jbc.M100758200. PMID 11402022.
- ^ a b c d e f Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF (Jul 2002). "ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer". Molecular Cell. 10 (1): 139–49. doi:10.1016/S1097-2765(02)00576-2. PMC 3516284. PMID 12150914.
- ^ a b c d e Ward A, Reyes CL, Yu J, Roth CB, Chang G (Nov 2007). "Flexibility in the ABC transporter MsbA: Alternating access with a twist". Proceedings of the National Academy of Sciences of the United States of America. 104 (48): 19005–10. Bibcode:2007PNAS..10419005W. doi:10.1073/pnas.0709388104. PMC 2141898. PMID 18024585.
- ^ a b Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA (Jun 2000). "Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily". Cell. 101 (7): 789–800. doi:10.1016/S0092-8674(00)80890-9. PMID 10892749. S2CID 18850076.
- ^ Fetsch EE, Davidson AL (Jul 2002). "Vanadate-catalyzed photocleavage of the signature motif of an ATP-binding cassette (ABC) transporter". Proceedings of the National Academy of Sciences of the United States of America. 99 (15): 9685–90. doi:10.1073/pnas.152204499. PMC 124977. PMID 12093921.
- ^ a b c d Reyes CL, Ward A, Yu J, Chang G (Feb 2006). "The structures of MsbA: Insight into ABC transporter-mediated multidrug efflux". FEBS Letters. 580 (4): 1042–8. Bibcode:2006FEBSL.580.1042R. doi:10.1016/j.febslet.2005.11.033. PMID 16337944. S2CID 34114828.
- ^ Ambudkar SV, Kim IW, Xia D, Sauna ZE (Feb 2006). "The A-loop, a novel conserved aromatic acid subdomain upstream of the Walker A motif in ABC transporters, is critical for ATP binding". FEBS Letters. 580 (4): 1049–55. Bibcode:2006FEBSL.580.1049A. doi:10.1016/j.febslet.2005.12.051. PMID 16412422. S2CID 20550226.
- ^ a b Geourjon C, Orelle C, Steinfels E, Blanchet C, Deléage G, Di Pietro A, Jault JM (Sep 2001). "A common mechanism for ATP hydrolysis in ABC transporter and helicase superfamilies". Trends in Biochemical Sciences. 26 (9): 539–44. doi:10.1016/S0968-0004(01)01907-7. PMID 11551790.
- ^ Ye J, Osborne AR, Groll M, Rapoport TA (Nov 2004). "RecA-like motor ATPases--lessons from structures". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1659 (1): 1–18. doi:10.1016/j.bbabio.2004.06.003. PMID 15511523.
- ^ a b Zaitseva J, Jenewein S, Jumpertz T, Holland IB, Schmitt L (Jun 2005). "H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB". The EMBO Journal. 24 (11): 1901–10. doi:10.1038/sj.emboj.7600657. PMC 1142601. PMID 15889153.
- ^ Maegley KA, Admiraal SJ, Herschlag D (Aug 1996). "Ras-catalyzed hydrolysis of GTP: a new perspective from model studies". Proceedings of the National Academy of Sciences of the United States of America. 93 (16): 8160–6. Bibcode:1996PNAS...93.8160M. doi:10.1073/pnas.93.16.8160. PMC 38640. PMID 8710841.
- ^ Matte A, Tari LW, Delbaere LT (Apr 1998). "How do kinases transfer phosphoryl groups?". Structure. 6 (4): 413–9. doi:10.1016/S0969-2126(98)00043-4. PMID 9562560.
- ^ a b Hollenstein K, Dawson RJ, Locher KP (Aug 2007). "Structure and mechanism of ABC transporter proteins". Current Opinion in Structural Biology. 17 (4): 412–8. doi:10.1016/j.sbi.2007.07.003. PMID 17723295.
- ^ a b c d e f g Higgins CF, Linton KJ (Oct 2004). "The ATP switch model for ABC transporters". Nature Structural & Molecular Biology. 11 (10): 918–26. doi:10.1038/nsmb836. PMID 15452563. S2CID 23058653.
- ^ Locher KP (Aug 2004). "Structure and mechanism of ABC transporters". Current Opinion in Structural Biology. 14 (4): 426–31. doi:10.1016/j.sbi.2004.06.005. PMID 15313236.
- ^ a b c d e f g h Oldham ML, Davidson AL, Chen J (Dec 2008). "Structural insights into ABC transporter mechanism". Current Opinion in Structural Biology. 18 (6): 726–33. doi:10.1016/j.sbi.2008.09.007. PMC 2643341. PMID 18948194.
- ^ a b c d Chang G (Nov 2003). "Multidrug resistance ABC transporters". FEBS Letters. 555 (1): 102–5. Bibcode:2003FEBSL.555..102C. doi:10.1016/S0014-5793(03)01085-8. PMID 14630327. S2CID 24228062.
- ^ Senior AE, al-Shawi MK, Urbatsch IL (Dec 1995). "The catalytic cycle of P-glycoprotein". FEBS Letters. 377 (3): 285–9. Bibcode:1995FEBSL.377..285S. doi:10.1016/0014-5793(95)01345-8. PMID 8549739. S2CID 20395778.
- ^ Simpson, Brent W.; Pahil, Karanbir S.; Owens, Tristan W.; Lundstedt, Emily A.; Davis, Rebecca M.; Kahne, Daniel; Ruiz, Natividad (20 August 2019). "Combining Mutations That Inhibit Two Distinct Steps of the ATP Hydrolysis Cycle Restores Wild-Type Function in the Lipopolysaccharide Transporter and Shows that ATP Binding Triggers Transport". mBio. 10 (4): e01931–19, /mbio/10/4/mBio.01931–19.atom. doi:10.1128/mBio.01931-19. PMC 6703430. PMID 31431556.
- ^ Martin C, Higgins CF, Callaghan R (Dec 2001). "The vinblastine binding site adopts high- and low-affinity conformations during a transport cycle of P-glycoprotein". Biochemistry. 40 (51): 15733–42. doi:10.1021/bi011211z. PMID 11747450.
- ^ Manciu L, Chang XB, Buyse F, Hou YX, Gustot A, Riordan JR, Ruysschaert JM (Jan 2003). "Intermediate structural states involved in MRP1-mediated drug transport. Role of glutathione". The Journal of Biological Chemistry. 278 (5): 3347–56. doi:10.1074/jbc.M207963200. PMID 12424247.
- ^ Kreimer DI, Chai KP, Ferro-Luzzi Ames G (Nov 2000). "Nonequivalence of the nucleotide-binding subunits of an ABC transporter, the histidine permease, and conformational changes in the membrane complex". Biochemistry. 39 (46): 14183–95. doi:10.1021/bi001066. PMID 11087367.
- ^ Vigano C, Margolles A, van Veen HW, Konings WN, Ruysschaert JM (Apr 2000). "Secondary and tertiary structure changes of reconstituted LmrA induced by nucleotide binding or hydrolysis. A fourier transform attenuated total reflection infrared spectroscopy and tryptophan fluorescence quenching analysis" (PDF). The Journal of Biological Chemistry. 275 (15): 10962–7. doi:10.1074/jbc.275.15.10962. PMID 10753896. S2CID 33274934.
- ^ Sonveaux N, Vigano C, Shapiro AB, Ling V, Ruysschaert JM (Jun 1999). "Ligand-mediated tertiary structure changes of reconstituted P-glycoprotein. A tryptophan fluorescence quenching analysis". The Journal of Biological Chemistry. 274 (25): 17649–54. doi:10.1074/jbc.274.25.17649. PMID 10364203.
- ^ Rosenberg MF, Velarde G, Ford RC, Martin C, Berridge G, Kerr ID, Callaghan R, Schmidlin A, Wooding C, Linton KJ, Higgins CF (Oct 2001). "Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle". The EMBO Journal. 20 (20): 5615–25. doi:10.1093/emboj/20.20.5615. PMC 125677. PMID 11598005.
- ^ a b Gilson, L.; Mahanty, H. K.; Kolter, R. (December 1990). "Genetic analysis of an MDR-like export system: the secretion of colicin V". The EMBO Journal. 9 (12): 3875–3884. doi:10.1002/j.1460-2075.1990.tb07606.x. ISSN 0261-4189. PMC 552155. PMID 2249654.
- ^ Choi, Young Hee; Yu, Ai-Ming (2014). "ABC Transporters in Multidrug Resistance and Pharmacokinetics, and Strategies for Drug Development". Current Pharmaceutical Design. 20 (5): 793–807. doi:10.2174/138161282005140214165212. ISSN 1381-6128. PMC 6341993. PMID 23688078.
- ^ McMurry L, Petrucci RE, Levy SB (Jul 1980). "Active efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli". Proceedings of the National Academy of Sciences of the United States of America. 77 (7): 3974–7. Bibcode:1980PNAS...77.3974M. doi:10.1073/pnas.77.7.3974. PMC 349750. PMID 7001450.
- ^ Rea PA (2007). "Plant ATP-binding cassette transporters". Annual Review of Plant Biology. 58: 347–75. doi:10.1146/annurev.arplant.57.032905.105406. PMID 17263663.
- ^ Bailly A, Yang H, Martinoia E, Geisler M, Murphy AS (2011). "Plant Lessons: Exploring ABCB Functionality Through Structural Modeling". Frontiers in Plant Science. 2: 108. doi:10.3389/fpls.2011.00108. PMC 3355715. PMID 22639627.
- ^ Geisler M, Murphy AS (Feb 2006). "The ABC of auxin transport: the role of p-glycoproteins in plant development". FEBS Letters. 580 (4): 1094–102. Bibcode:2006FEBSL.580.1094G. doi:10.1016/j.febslet.2005.11.054. PMID 16359667. S2CID 23368914.
- ^ a b c d Yang H, Murphy AS (Jul 2009). "Functional expression and characterization of Arabidopsis ABCB, AUX 1 and PIN auxin transporters in Schizosaccharomyces pombe". The Plant Journal. 59 (1): 179–91. doi:10.1111/j.1365-313X.2009.03856.x. PMID 19309458.
- ^ Blakeslee JJ, Peer WA, Murphy AS (Oct 2005). "Auxin transport". Current Opinion in Plant Biology. 8 (5): 494–500. Bibcode:2005COPB....8..494B. doi:10.1016/j.pbi.2005.07.014. PMID 16054428.
- ^ Kretzschmar T, Burla B, Lee Y, Martinoia E, Nagy R (Sep 2011). "Functions of ABC transporters in plants" (PDF). Essays in Biochemistry. 50 (1): 145–60. doi:10.1042/bse0500145. PMID 21967056.
- ^ Kubeš M, Yang H, Richter GL, Cheng Y, Młodzińska E, Wang X, Blakeslee JJ, Carraro N, Petrášek J, Zažímalová E, Hoyerová K, Peer WA, Murphy AS (Feb 2012). "The Arabidopsis concentration-dependent influx/efflux transporter ABCB4 regulates cellular auxin levels in the root epidermis". The Plant Journal. 69 (4): 640–54. doi:10.1111/j.1365-313X.2011.04818.x. PMID 21992190.
- ^ Dawson RJ, Locher KP (Mar 2007). "Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP". FEBS Letters. 581 (5): 935–8. Bibcode:2007FEBSL.581..935D. doi:10.1016/j.febslet.2007.01.073. PMID 17303126. S2CID 19960736.
- ^ Velamakanni S, Yao Y, Gutmann DA, van Veen HW (Sep 2008). "Multidrug transport by the ABC transporter Sav1866 from Staphylococcus aureus". Biochemistry. 47 (35): 9300–8. doi:10.1021/bi8006737. PMID 18690712.
- ^ Reuter G, Janvilisri T, Venter H, Shahi S, Balakrishnan L, van Veen HW (Sep 2003). "The ATP binding cassette multidrug transporter LmrA and lipid transporter MsbA have overlapping substrate specificities". The Journal of Biological Chemistry. 278 (37): 35193–8. doi:10.1074/jbc.M306226200. PMID 12842882.
- ^ Raetz CR, Reynolds CM, Trent MS, Bishop RE (2007). "Lipid A modification systems in gram-negative bacteria". Annual Review of Biochemistry. 76: 295–329. doi:10.1146/annurev.biochem.76.010307.145803. PMC 2569861. PMID 17362200.
- ^ Chang G, Roth CB (Sep 2001). "Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters". Science. 293 (5536): 1793–800. Bibcode:2001Sci...293.1793C. doi:10.1126/science.293.5536.1793. PMID 11546864. (Retracted, see doi:10.1126/science.314.5807.1875b, PMID 17185584) (Retracted, see doi:10.1126/science.314.5807.1875b)
- ^ Reyes CL, Chang G (May 2005). "Structure of the ABC transporter MsbA in complex with ADP.vanadate and lipopolysaccharide". Science. 308 (5724): 1028–31. Bibcode:2005Sci...308.1028R. doi:10.1126/science.1107733. PMID 15890884. S2CID 37250061. (Retracted, see doi:10.1126/science.314.5807.1875b, PMID 17185584) (Retracted, see doi:10.1126/science.314.5807.1875b)
- ^ Buchaklian AH, Funk AL, Klug CS (Jul 2004). "Resting state conformation of the MsbA homodimer as studied by site-directed spin labeling". Biochemistry. 43 (26): 8600–6. doi:10.1021/bi0497751. PMID 15222771.
- ^ a b c Dong J, Yang G, McHaourab HS (May 2005). "Structural basis of energy transduction in the transport cycle of MsbA". Science. 308 (5724): 1023–8. Bibcode:2005Sci...308.1023D. doi:10.1126/science.1106592. PMID 15890883. S2CID 1308350.
- ^ Borbat PP, Surendhran K, Bortolus M, Zou P, Freed JH, Mchaourab HS (Oct 2007). "Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis". PLOS Biology. 5 (10): e271. doi:10.1371/journal.pbio.0050271. PMC 2001213. PMID 17927448.
- ^ Zhang, Ge; Baidin, Vadim; Pahil, Karanbir S.; Moison, Eileen; Tomasek, David; Ramadoss, Nitya S.; Chatterjee, Arnab K.; McNamara, Case W.; Young, Travis S.; Schultz, Peter G.; Meredith, Timothy C.; Kahne, Daniel (7 May 2018). "Cell-based screen for discovering lipopolysaccharide biogenesis inhibitors". Proceedings of the National Academy of Sciences. 115 (26): 6834–6839. Bibcode:2018PNAS..115.6834Z. doi:10.1073/pnas.1804670115. PMC 6042065. PMID 29735709.
- ^ Ho, Hoangdung; Miu, Anh; Alexander, Mary Kate; Garcia, Natalie K.; Oh, Angela; Zilberleyb, Inna; Reichelt, Mike; Austin, Cary D.; Tam, Christine; Shriver, Stephanie; Hu, Huiyong; Labadie, Sharada S.; Liang, Jun; Wang, Lan; Wang, Jian; Lu, Yan; Purkey, Hans E.; Quinn, John; Franke, Yvonne; Clark, Kevin; Beresini, Maureen H.; Tan, Man-Wah; Sellers, Benjamin D.; Maurer, Till; Koehler, Michael F. T.; Wecksler, Aaron T.; Kiefer, James R.; Verma, Vishal; Xu, Yiming; Nishiyama, Mireille; Payandeh, Jian; Koth, Christopher M. (May 2018). "Structural basis for dual-mode inhibition of the ABC transporter MsbA". Nature. 557 (7704): 196–201. Bibcode:2018Natur.557..196H. doi:10.1038/s41586-018-0083-5. PMID 29720648. S2CID 13660653.
- ^ Gutmann DA, Ward A, Urbatsch IL, Chang G, van Veen HW (Jan 2010). "Understanding polyspecificity of multidrug ABC transporters: closing in on the gaps in ABCB1". Trends in Biochemical Sciences. 35 (1): 36–42. doi:10.1016/j.tibs.2009.07.009. PMC 4608440. PMID 19819701.
- ^ Leonard GD, Fojo T, Bates SE (2003). "The role of ABC transporters in clinical practice". The Oncologist. 8 (5): 411–24. doi:10.1634/theoncologist.8-5-411. PMID 14530494. S2CID 2780630.
- ^ Lage L (2009). "ABC Transporters as Target for RNA Interference-mediated Reversal of Multidrug Resistance". ABC Transporters in Microorganisms. Caister Academic. ISBN 978-1-904455-49-3.
- ^ Annaert PP, Turncliff RZ, Booth CL, Thakker DR, Brouwer KL (Oct 2001). "P-glycoprotein-mediated in vitro biliary excretion in sandwich-cultured rat hepatocytes". Drug Metab Dispos. 29 (10): 1277–83. PMID 11560870.
- ^ a b Annaert PP, Brouwer KL (Mar 2005). "Assessment of drug interactions in hepatobiliary transport using rhodamine 123 in sandwich-cultured rat hepatocytes". Drug Metab Dispos. 33 (3): 388–94. doi:10.1124/dmd.104.001669. PMID 15608134. S2CID 7063502.
- ^ Matsson, Pär (2007). "ATP-Binding Cassette Efflux Transporters and Passive Membrane Permeability in Drug Absorption and Disposition". Diva.
- ^ Glavinas H, Krajcsi P, Cserepes J, Sarkadi B (Jan 2004). "The role of ABC transporters in drug resistance, metabolism and toxicity". Current Drug Delivery. 1 (1): 27–42. doi:10.2174/1567201043480036. PMID 16305368.
- ^ Glavinas H, Méhn D, Jani M, Oosterhuis B, Herédi-Szabó K, Krajcsi P (Jun 2008). "Utilization of membrane vesicle preparations to study drug-ABC transporter interactions". Expert Opinion on Drug Metabolism & Toxicology. 4 (6): 721–32. doi:10.1517/17425255.4.6.721. PMID 18611113. S2CID 86198612.
- ^ This entire volume is dedicated to various methods used: Nikaido H, Hall J (1998). ABC Transporters: Biochemical, Cellular, and Molecular Aspects. Methods in Enzymology. Vol. 292. pp. 3–853. doi:10.1016/s0076-6879(00)x0188-7. ISBN 9780121821937. PMID 9711542.
- ^ Horio M, Gottesman MM, Pastan I (May 1988). "ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells". Proceedings of the National Academy of Sciences of the United States of America. 85 (10): 3580–4. Bibcode:1988PNAS...85.3580H. doi:10.1073/pnas.85.10.3580. PMC 280257. PMID 3368466.
- ^ Vasiliou, V; Vasiliou, K; Nebert, DW (April 2009). "Human ATP-binding cassette (ABC) transporter family". Human Genomics. 3 (3): 281–90. doi:10.1186/1479-7364-3-3-281. PMC 2752038. PMID 19403462.
- ^ a b Chen ZS, Tiwari AK (Sep 2011). "Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases". The FEBS Journal. 278 (18): 3226–45. doi:10.1111/j.1742-4658.2011.08235.x. PMC 3168698. PMID 21740521.
- ^ Saier MH (Jun 2000). "A functional-phylogenetic classification system for transmembrane solute transporters". Microbiology and Molecular Biology Reviews. 64 (2): 354–411. doi:10.1128/MMBR.64.2.354-411.2000. PMC 98997. PMID 10839820.; Saier Lab Bioinformatics Group. "3.A.1 The ATP-binding Cassette (ABC) Superfamily". Transporter Classification Database (TCDB). University of California San Diego.
- ^ Khwaja M, Ma Q, Saier MH (Mar 2005). "Topological analysis of integral membrane constituents of prokaryotic ABC efflux systems". Research in Microbiology. 156 (2): 270–7. doi:10.1016/j.resmic.2004.07.010. PMID 15748994.
- ^ Zheng, WH; Västermark, Å; Shlykov, MA; Reddy, V; Sun, EI; Saier MH, Jr (6 May 2013). "Evolutionary relationships of ATP-Binding Cassette (ABC) uptake porters". BMC Microbiology. 13: 98. doi:10.1186/1471-2180-13-98. PMC 3654945. PMID 23647830.
Further reading
[edit]- Szentpétery Z, Kern A, Liliom K, Sarkadi B, Váradi A, Bakos E (Oct 2004). "The role of the conserved glycines of ATP-binding cassette signature motifs of MRP1 in the communication between the substrate-binding site and the catalytic centers". The Journal of Biological Chemistry. 279 (40): 41670–8. doi:10.1074/jbc.M406484200. PMID 15252017.
- Fitzgerald ML, Okuhira K, Short GF, Manning JJ, Bell SA, Freeman MW (Nov 2004). "ATP-binding cassette transporter A1 contains a novel C-terminal VFVNFA motif that is required for its cholesterol efflux and ApoA-I binding activities". The Journal of Biological Chemistry. 279 (46): 48477–85. doi:10.1074/jbc.M409848200. PMID 15347662.
- Linton KJ (2011). The ABC Transporters of Human Physiology and Disease: The Genetics and Biochemistry of ATP Binding Cassette. World Scientific. ISBN 978-981-4280-06-8. Archived from the original on 2012-05-11. Retrieved 2012-05-16.
External links
[edit]- Classification of ABC transporters in TCDB
- ABCdb Archaeal and Bacterial ABC Systems database, ABCdb
- ATP-Binding+cassette+transporters at the U.S. National Library of Medicine Medical Subject Headings (MeSH)