ISSN: 1838-7640Theranostics
- Current issue
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Volume 11; 2021
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Top
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
Global reach, higher impact
Theranostics 2019; 9(9):2526-2540. doi:10.7150/thno.32915 This issue Cite
Research Paper
Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit
Chih-Chia Kuo1,2*, Hsiang-Hsi Ling1,2*, Ming-Chen Chiang1,2*, Chu-Hung Chung1,2, Wen-Ying Lee3, Cheng-Ying Chu4, Yu-Chih Wu1,5, Cheng-Hsun Chen1, Yi-Wen Lai1, I-Lin Tsai1,2, Chia-Hsiung Cheng1,2, Cheng-Wei Lin1,2,5
1. Department of Biochemistry and Molecular Cell Biology, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan.
2. Graduate Institute of Medical Sciences, College of Medicine, Taipei Medical University, Taipei, Taiwan.
3. Department of Cytopathology, Chi Mei Medical Center, Tainan, Taiwan.
4. TMU Research Center of Cancer Translational Medicine, Taipei Medical University, Taipei, Taiwan.
5. Center for Cell Therapy and Regeneration Medicine, Taipei Medical University, Taipei, Taiwan.
*Chih-Chia Kuo, Hsiang-Hsi Ling, and Ming-Chen Chiang contributed equally to this work
Citation:
Kuo CC, Ling HH, Chiang MC, Chung CH, Lee WY, Chu CY, Wu YC, Chen CH, Lai YW, Tsai IL, Cheng CH, Lin CW. Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit. Theranostics 2019; 9(9):2526-2540. doi:10.7150/thno.32915. https://www.thno.org/v09p2526.htm
Other stylesAbstract
Rationale: Cancer cells reprogram cellular metabolism to fulfill their needs for rapid growth and metastasis. However, the mechanism controlling this reprogramming is poorly understood. We searched for upregulated signaling in metastatic colorectal cancer and investigated the mechanism by which Glut3 promotes tumor metastasis.
Methods: We compared RNA levels and glycolytic capacity in primary and metastatic colon cancer. The expression and association of Glut3 with clinical prognosis in colon cancer tissues was determined by immunohistochemistry. Glut3 gain-of-function and loss-of-function were established using colon cancer HCT116, HT29, and metastatic 116-LM cells, and tumor invasiveness and stemness properties were evaluated. Metabolomic profiles were analyzed by GC/MS and CE-TOF/MS. The metastatic burden in mice fed a high-fat sucrose diet was assessed by intravenous inoculation with Glut3 knockdown 116-LM cells.
Results: Upregulation of glycolytic genes and glycolytic capacity was detected in metastatic colorectal cancer cells. Specifically, Glut3 overexpression was associated with metastasis and poor survival in colorectal cancer patients. Mechanistically, Glut3 promoted invasiveness and stemness in a Yes-associated protein (YAP)-dependent manner. Activation of YAP in turn transactivated Glut3 and regulated a group of glycolytic genes. Interestingly, the expression and phosphorylation of PKM2 were concomitantly upregulated in metastatic colorectal cancer, and it was found to interact with YAP and enhance the expression of Glut3. Importantly, a high-fat high-sucrose diet promoted tumor metastasis, whereas the inhibition of either Glut3 or YAP effectively reduced the metastatic burden.
Conclusion: Activation of the Glut3-YAP signaling pathway acts as a master activator to reprogram cancer metabolism and thereby promotes metastasis. Our findings reveal the importance of metabolic reprogramming in supporting cancer metastasis as well as possible therapeutic targets.
Keywords: cancer metabolism, colorectal cancer, glucose transporter, metastasis, YAP
Introduction
Colorectal cancer (CRC) is the third most common cancer and the second most common fatal cancer worldwide. Approximately 25% of patients experience systemic metastases, and the most frequently metastatic sites are the liver and lung [1]. Despite major research efforts and clinical progress, predictable biomarkers for colorectal cancer metastasis and therapeutics are not yet fully understood. Recently, alterations in cellular metabolism have been recognized as emerging hallmarks of cancer, and the most appreciated hallmark is aerobic glycolysis, also known as the Warburg effect [2]. It is believed that cancer cells increase glucose consumption to meet the high energy and macromolecule demands required for rapid growth [3], and several studies have reported that oncogenic signals drive the malignant progression of cancer cells by modulating metabolic enzymes. For instance, the oncogene c-Myc transactivates most glycolytic genes, including glucose transporter 1 (Glut1) and lactate dehydrogenase A (LDHA) [4, 5]. Hypoxia-inducing factor 1 (HIF-1) regulates LDHA and pyruvate dehydrogenase kinase isozyme 1 (PDK1) [6]. Other recent evidence has shown that mitogen signaling promotes tumor growth by modulating pyruvate kinase M2 (PKM2) [7]. These evidence indicate that the regulatory mechanisms underlying cellular metabolism in cancer cells are far more complex than previously believed and warrant investigation.
The glucose transporter (encoded by SLC2A) family comprises enzymes involved in the first step of glycolysis and facilitates glucose entry into cells. The Glut family includes 14 members that transport glucose or other substances in different tissues [8]. Glut1 is ubiquitously expressed in normal tissues and is overexpressed in many cancer types. In contrast to Glut1, Glut3 is expressed in neuronal cells but is less expressed in other organs under normal conditions. Interestingly, recent studies have reported the elevated expression of Glut3 in glioma and hepatoblastoma [9]. The induction of Glut3 facilitates metabolic adaptation to nutrient deprivation in brain tumor-initiating cells and is responsible for TGFβ-induced EMT in nonsmall cell lung cancer [10, 11]. However, the regulation of Glut3 in CRC metastasis and the contribution of Glut3 to this process are largely unknown.
In an attempt to identify biomarkers for metastatic CRC, we investigated transcriptome in primary and metastatic CRC tissues. Interestingly, we found that Glut3 expression and glycolytic capacity were concomitantly increased in metastatic CRC. We further demonstrated that Glut3 operates through a positive regulatory loop by collaborating with the Hippo cascade transducer YAP. We show that Glut3 is responsible for the activation of YAP, which in turn transactivates glycolytic genes, including Glut3 itself. Moreover, oncogenic signal promotes the association of YAP and PKM2, thereby enhancing Glut3 expression and promoting tumor invasiveness. Blockade of Glut3 and YAP effectively represses CRC aggressiveness and metastasis.
Results
Overexpression of Glut3 is an independent marker for metastatic CRC
To identify genes associated with CRC metastasis, we analyzed the RNA level in 7 pairs of primary and liver metastatic CRC tissues. Interestingly, gene ontology annotation revealed that cellular metabolic processes were altered. In particular, we found that the expression of Glut3 was upregulated in metastatic tumor tissues (Figure 1A and B). Transcriptome sequencing data from TCGA revealed that Glut3 overexpression was closely associated with the aggressive clinicopathological features of CRC (Figure 1C and Table S1). Accordingly, CRC patients with high Glut3 expression were associated with low overall and recurrence-free survival rates (Figure 1D). Immunohistochemistry analysis results confirmed that Glut3 protein was increased in metastatic CRC tissues compared with primary tumors in both paired and unpaired samples (Figure 1E). Intriguingly, data retrieved from TCGA revealed that the correlation between Glut3 and poor survival was found more often in colon and lung squamous cancers than in other cancer types (Figure 1F). Similar results from other independent cohorts also showed that Glut3 overexpression correlated with poor outcomes, particularly in colon and lung cancers (Figure S1A).
To better understand the involvement of the Glut family in CRC, we analyzed the expression of Glut isoforms 1-4. The results showed that the expression of Glut1 and Glut3, but not that of Glut2 and Glut4, was upregulated in tumors compared with normal tissues (Figure S1B and S1C). However, expression of Glut1 was associated only with tumor stage (Figure S1D). Overall and recurrence-free survival was not correlated with Glut1 expression (Figure S1E). These data indicate that Glut3 expression is an indispensable marker for CRC.
Aerobic glycolysis is upregulated in metastatic CRC
To gain insights into the role of cellular metabolism in CRC metastasis, we examined the bioenergetic profile in colorectal cancer HCT116 cells and their metastatic derivatives, 116-LM cells, which were obtained from pulmonary metastasis of HCT116 cells [12]. The increased migration and invasion in 116-LM cells was confirmed by Transwell assay (Figure S2A). Importantly, 116-LM cells exhibited an increased glycolytic rate and glycolytic capacity (Figure 1G). Quantitative PCR analysis revealed that the expression of glycolysis-related genes was increased in 116-LM cells (Figure 1H). Notably, the expression of Glut3 rather than that of other isoforms was drastically elevated in 116-LM cells compared with the parental HCT116 cells (Figure 1H). Western blot results confirmed that Glut3 protein levels were upregulated in 116-LM cells (Figure S2B). In contrast, the protein level of Glut1 was similar to that of Glut3 to some extent in colon cancer cell lines, but the Glut1 protein levels between HCT116 and 116-LM cells were not changed (Figure S2B).
Figure 1
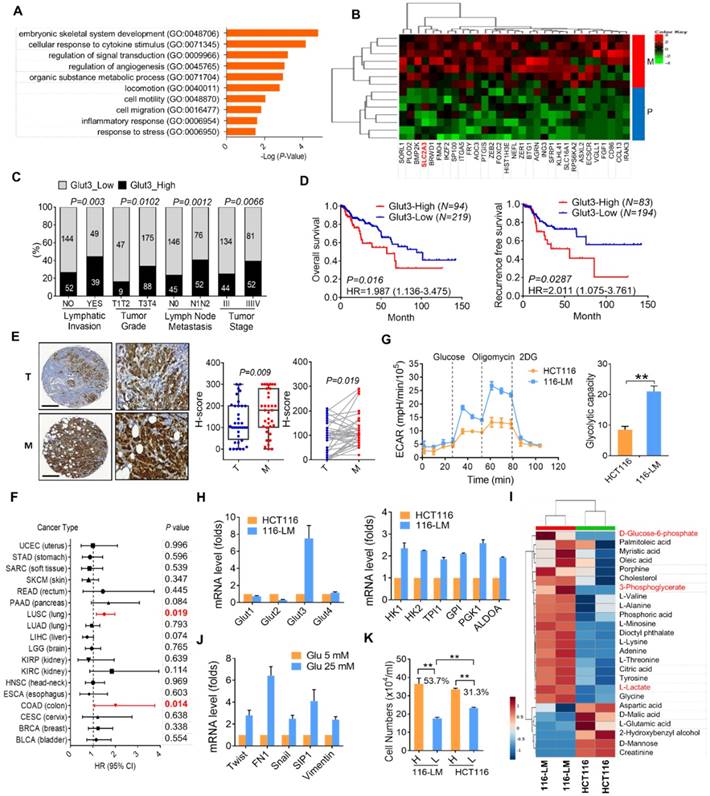
Upregulation of Glut3 and its correlation with poor survival in CRC patients. (A) Gene ontology annotation shows an altered signaling pathway in primary and liver metastatic CRC. (B) Heatmap representation of upregulated genes associated with metabolic process. P: primary tumor; M: liver metastatic tumor. (C and D) High Glut3 expression was associated with lymphatic invasion, tumor grade, lymph node metastasis, tumor stage (C), and poor survival prognosis (D) in colorectal cancer patients using the TCGA dataset (COADREAD). Clinicopathological correlation was examined by a chi-square test, and survival prognosis was calculated by Kaplan-Meier analysis. (E) Representative IHC images of Glut3 protein expression in primary and metastatic colon cancer tissues. Scale bar=300 μm. Enlarged pictures are shown in the right panel. Unpaired t-tests were used to compare H-score analyses of primary and metastatic tumor tissues. A paired t-test was used to compare H-score analyses of paired tissue samples. (F) Overexpression of Glut3 was associated with poor prognosis for colon cancer (COAD) and lung squamous cell carcinoma (LUSC) in the TCGA database. HR=hazard ratio. The P value was determined by the log-rank test. (G) Increased glycolytic capacity in metastatic 116-LM cells. Real-time monitored glycolytic activity was determined by measuring the extracellular acidification rate (ECAR). Data are presented as the mean ± SE (left). The total cellular glycolytic capacity in 116-LM and HCT116 cells (right) is shown. ** P<0.01 versus the capacity in HCT116 cells. (H) Quantitative PCR analysis of glycolytic gene expression in the HCT116 and 116-LM cells. Data are representative of three independent experiments, and expression levels relative to those in HCT116 cells are shown. (I) Heatmap representation of cellular metabolites determined by GC/MS analysis in 116-LM and HCT116 cells. Data were obtained from two biological repeats. The glycolysis products are shown in red. (J) 116-LM cells were exposed to low (5 mM) or high (25 mM) concentrations of glucose for 48 h, and the expression of EMT genes was analyzed by quantitative PCR. (K) 116-LM and HCT116 cells (105/ml) were cultured in complete medium and changed to medium with low (5 mM) or high (25 mM) concentrations of glucose for 48 h. Cell number was measured by trypan blue exclusion assay. ** P<0.01 compared to the indicated group.
To further examine cellular metabolites in 116-LM cells, GC/MS was performed, and 43 metabolites were identified. The results showed that metastatic 116-LM cells increased glycolysis products, including glucose-6-phosphate, 3-phosphoglycerate, and lactate (Figure 1I), which is correlated with the upregulation of glycolytic capacity indicated by the ECAR (Figure 1G). Interestingly, the levels of amino acids and fatty acids were also upregulated in 116-LM cells, suggesting a demand for protein and macromolecule syntheses in metastatic cells (Figure 1I). To evaluate the contribution of upregulated glycolysis in 116-LM cells, cells were treated with high or low concentrations of glucose. Quantitative PCR analysis showed that high glucose promoted the expression of EMT and glycolytic genes (Figure 1J and Figure S2D). In contrast, the inhibition of glycolysis by 2-deoxyglucose (2DG) suppressed EMT genes (Figure S2E). Moreover, the growth of 116-LM cells was more vulnerable to glucose restriction than the growth of HCT116 cells (Figure 1K). Low glucose treatment decreased the growth of 116-LM and HCT116 cells by 53% and 31% from their levels in high glucose, respectively (P<0.01). Together, these data suggest that metastatic CRC requires greater glucose utilization.
Glut3 promotes CRC aggressiveness and stemness through a YAP-dependent pathway
To determine the contribution of Glut3 to CRC metastasis, we generated Glut3-overexpressing HCT116 and Glut3-silencing 116-LM cells (Figure S2C). Glut3 overexpression increased the transcription of EMT genes and promoted cell migration and invasion (Figure 2A and 2B). Additionally, Glut3 overexpression increased the transcription of stemness-related genes (Figure 2C) and the formation of tumorspheres (Figure 2E), properties associated with cancer stem cells. However, this phenomenon was conversely reduced in Glut3 knockdown cells (Figure 2D and 2E). Knockdown of Glut3 also reduced the promoter activity of stemness-related transcription factors (Figure 2F). Moreover, knockout of Glut3 using the CRISPR/Cas9 systems significantly reduced cell migration and invasion as well as tumorsphere formation (Figure 2G and Figure S3A and S3B). These data indicate that Glut3 is crucial for the invasiveness and stemness properties of metastatic CRC.
To study the mechanism underlying the promotion of CRC invasiveness and stemness by Glut3, we examined the Hippo pathway, as it has been reported to be regulated by cellular energy status and has drawn increasing attention for its role in tumor promotion. Moreover, we previously identified that the expression of YAP is upregulated and plays a role in the invasive properties of 116-LM cells [12]. Thus, we investigated the relationship between Glut3 and Hippo signaling. Consistently, 116-LM cells exhibited increased levels of YAP compared with the parental HCT116 cells (Figure S2F), and treatment with glucose reduced YAP phosphorylation and increased protein level of YAP and its paralog TAZ (Figure S2G). In contrast, blocking ATP production by the addition of 2DG induced YAP phosphorylation, suppressed YAP/TAZ level (Figure S2G) and subsequently inhibited the expression of YAP downstream genes (Figure 2H). Moreover, low glucose concentrations (5 mM) robustly increased YAP protein levels in 116-LM cells, while YAP was equivalently induced by high glucose concentrations (25 mM) in the parental cells (Figure 2H). We hypothesized that metastatic CRC are more susceptible to glucose due to elevated levels of Glut3. Indeed, the release of glucose induced YAP upregulation in control knockdown 116-LM cells, while Glut3 silencing remarkably diminished YAP in either glucose starvation or glucose-releasing conditions (Figure 2I). Moreover, Glut3 silencing remarkably suppressed the invasiveness of 116-LM cells compared with HCT116 cells (Figure 2J and Figure S2I), indicating that metastatic CRC is addicted to Glut3, possibly through the activation of YAP signaling.
To further determine whether Glut3 is responsible for YAP activation and therefore promotes CRC metastasis, we examined the expression of YAP target genes. Quantitative PCR assays revealed that the YAP target genes were upregulated in Glut3-overexpressing HCT116 cells (Figure 2K), while they were downregulated in Glut3-silenced 116-LM cells (Figure S2J). YAP facilitates the regulation of downstream gene expression by binding with TEAD transcription factors. We further found that Glut3 silencing repressed the activity of a TEAD-responsive luciferase reporter (Figure 2L). Additionally, the increased capacities for tumor migration/invasion and tumorsphere formation as a result of Glut3 overexpression were suppressed by YAP silencing (Figure 2M). Gene set enrichment analysis (GSEA) showed that expression of Glut3 was significantly associated with the YAP signature in CRC patients (Figure 2N and Figure S4A). These findings indicate that the effects of Glut3 on CRC aggressiveness and stemness are dependent on YAP.
Figure 2

Glut3 promotes aggressiveness and stemness through YAP. (A) Quantitative PCR analysis of expression of the EMT genes in HCT116 cells stably overexpressing Glut3. (B) Tumor migration and invasion ability in mock- and Glut3-overexpressing HCT116 cells. Representative images are presented (left panel), and the relative percentages of migratory and invasive cells were counted (right panel). (C and D) Quantitative PCR analysis of the expression of stemness genes in HCT116 cells stably overexpressing Glut3 (C) or in Glut3-silenced HT29 cells (D). (E) Tumorsphere formation in Glut3-overexpressing HCT116 or Glut3-silenced HT29 cells. Representative images are presented (left panel), and the number of tumorspheres was counted (right panel). Scale bar=100 μm. (F) Luciferase reporter assay of stemness-related transcription factors in 116-LM/shRFP and 116-LM/shGlut3 cells. (G) Knockout of Glut3 impeded migration (a), invasion (b) and tumorsphere formation (c) in 116-LM cells. Representative images are presented (left panel), and quantitative data are shown (right panel). Scale bar=100 μm. (H) 116-LM and HCT116 cells were maintained in glucose-free medium for 8 h, followed by treatment with glucose for an additional 2 h. YAP expression was measured by Western blot. (I) 116-LM/shRFP and 116-LM/shGlut3 cells were starved with glucose deprivation or released by glucose (25 mM) treatment for an additional 2 h. Protein expression was analyzed by Western blot. (J) Comparison of cell invasion in Glut3-silenced HCT116 and 116-LM cells. (K) Quantitative PCR analysis of YAP downstream genes in Glut3-overexpressing HCT116 cells. (L) Knockdown of Glut3 inhibited the transcriptional activity of YAP, as measured by a TEAD luciferase reporter assay. (M) Knockdown of YAP abrogated tumor invasiveness and stemness in Glut3-overexpressing HCT116 cells. Representative images of migration (a), invasion (b), and tumorsphere formation (c) are presented (left panel), and quantitative data (right panel) are shown. Scale bar=100 μm. (N) GSEA shows that Glut3 is associated with the YAP signature. NES: net enrichment score. The results are depicted as the mean ± SEM. * P <0.05, ** P <0.01, determined by unpaired two-tailed Student's t-test.
Glut3-YAP signaling loops are crucial for CRC malignancy
Our aforementioned data show that Glut3 regulates YAP signaling. Interestingly, a recent study reported that inhibition of YAP repressed Glut3 expression in brain tumors [13], suggesting that Glut3 might be a YAP downstream target. However, the detailed underlying mechanism is not yet understood. Thus, we further examined the relationship between Glut3 and YAP. Quantitative PCR assays showed that YAP silencing downregulated Glut3 mRNA levels in both 116-LM and HT29 cells (Figure 3A and Figure S5A and S5B). We also found that knockdown of YAP decreased the expression of glycolysis genes (Figure 3A and Figure S5A and S5B). Conversely, overexpression of YAP5SA, a constitutively active YAP mutant, induced the mRNA expression of Glut3 and glycolysis genes in HCT116 cells (Figure 3A). Accordingly, addition of a TEAD inhibitor, verteporfin, suppressed Glut3 production (Figure S5C). Moreover, ChIP assay revealed YAP binding in the -1164/-1155 region of the Glut3 promoter (Figure 3B), and YAP expression induced Glut3 promoter activity (Figure 3C and Figure S5D). However, the deletion of the -1164/-1155 region diminished YAP-elicited transactivation of Glut3 (Figure 3C). Similarly, inhibition of YAP downregulated the promoter activity of Glut3 (Figure 3D), whereas overexpression of YAP5SA restored the production of Glut3 (Figure 3E). Furthermore, the reduced migration/invasion and tumorsphere formation in Glut3-silenced 116-LM cells were rescued upon reintroduction of YAP (Figure 3F). TCGA data further confirmed the positive correlation between Glut3 and YAP/TAZ mRNA in CRC patients (Figure 3G and Figure S4B) and showed that patients who coexpressed Glut3 with YAP/TAZ had the worst survival probability (Figure 3H and Figure S4C).
Because the inhibition of ATP production by 2DG suppressed YAP activity (Figure S2F), we wanted to determine whether inhibiting glycolysis impaired Glut3 expression. The results showed that inhibition of YAP by 2DG was accompanied by a reduction in Glut3 at both the mRNA and protein levels (Figure S5E and S5F). However, the inhibition of Glut3 by 2DG was rescued by YAP (Figure S5G). Altogether, these data support the observation of concomitant elevations in YAP and Glut3 in metastatic 116-LM cells (Figure S2E) and show that the reciprocal activation between Glut3 and YAP signaling acts as a feedback loop that fosters glucose-mediated CRC metastasis.
Figure 3
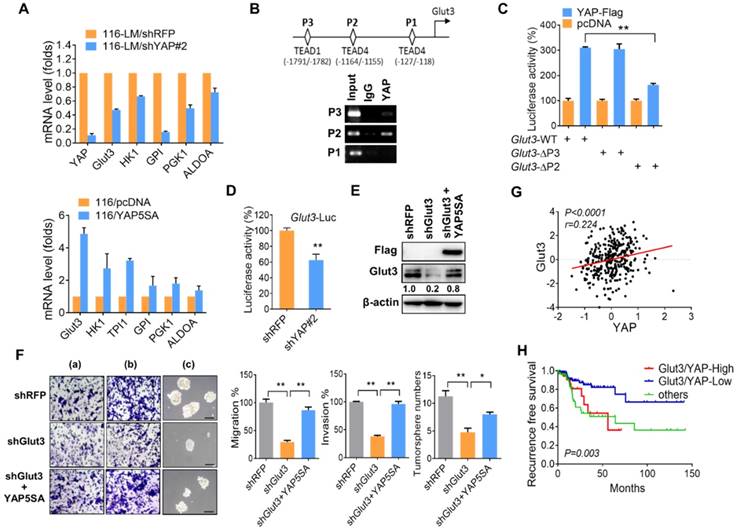
YAP transcriptionally regulates Glut3. (A) Quantitative PCR analysis of the expression of the glycolytic genes in CRC in which YAP is stably overexpressed (lower panel) or silenced (upper panel). (B) Illustration of three putative TEAD binding sites in the Glut3 promoter (upper panel). Chromatin immunoprecipitation analysis of YAP binding three tentative binding sites of the Glut3 promoter (lower panel). (C) 293T cells were cotransfected with YAP and deletion mutations of the Glut3 promoter. The transcriptional activity of Glut3 was determined by luciferase reporter assay. (D) Knockdown of YAP in 116-LM cells inhibited transcription of Glut3, as determined by luciferase reporter assay. (E) Overexpression of YAP5SA rescued Glut3 levels, and protein expression was analyzed by Western blot. (F) Overexpression of YAP5SA restored the capacities of 116-LM/Glut3 knockdown cells for invasiveness and stemness. Representative images of migration (a), invasion (b), and tumorsphere formation (c) are presented (upper panel), and quantitative data (lower panel) are shown. Scale bar=100 μm. The results are depicted as the mean ± SEM. * P <0.05, ** P <0.01, determined by unpaired two-tailed Student's t-test. (G) Pearson correlation analysis of YAP and Glut3 mRNA in the TCGA COADREAD dataset. (H) Kaplan-Meier curve shows that YAP and Glut3 coexpression results in a poor survival probability in colon cancer patients.
The association of YAP with PKM2 enhances Glut3 expression
Increasing evidence has shown that PKM2 acts as a key molecule in linking the reprogramming of cancer cell metabolism and oncogene-mediated tumorigenesis. Nonphosphorylated PKM2 exhibits pyruvate kinase activity, whereas phosphorylated PKM2 translocates to the nucleus and enhances the expression of oncogenic genes. Interestingly, it has been reported that the phosphorylation of PKM2 facilitates nuclear translocation through interaction with the WW domain of PIN1 protein [7]. Because YAP possesses a WW domain, we therefore investigated the relationship between YAP and PKM2. We found that the total and phosphorylated PKM2 were increased in metastatic 116-LM cells compared with HCT116 cells (Figure 4A). Moreover, phosphorylation of PKM2 was increased in the nuclear fraction of 116-LM cells (Figure 4A); thus, we explored whether increased Glut3 and/or YAP has an effect on PKM2 in metastatic CRC. The results showed that Glut3 silencing reduced total PKM2, as well as PKM2 phosphorylation to some extent, compared with the YAP knockdown cells, which robustly suppressed the expression and phosphorylation of PKM2 (Figure S6A). Moreover, YAP suppression decreased the presence of nuclear PKM2, whereas the ectopic overexpression of YAP5SA induced the nuclear translocation of PKM2 (Figure 4B and 4C). We also found that suppression of YAP decreased PKM2 protein stability (Figure 4D).
Figure 4
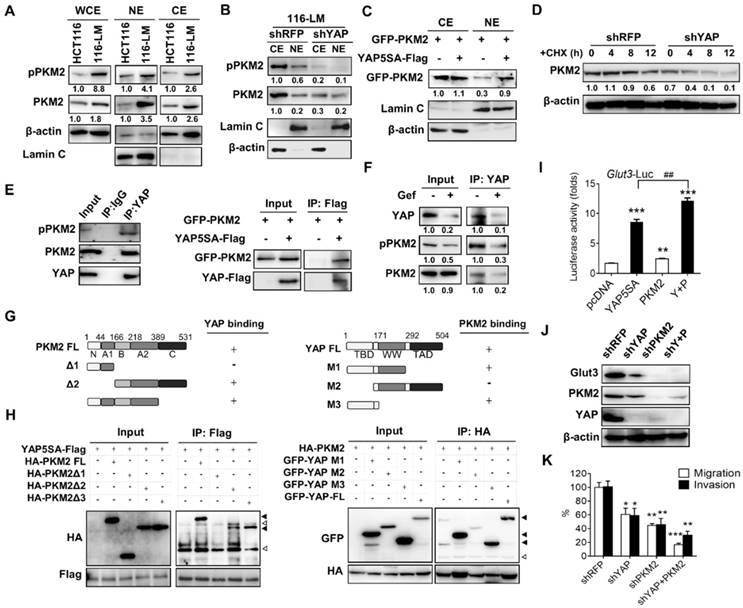
PKM2 interacts with YAP and enhances Glut3 expression. (A) Western blot analysis of total PKM2 and phosphorylated PKM2 levels in the whole-cell extract (WCE) and nuclear extract (NE) of HCT116 and 116-LM cells. (B) Western blot analysis of total PKM2 and phosphorylated PKM2 levels in the cytosolic extract (CE) and nuclear extract (NE) of 116-LM/shRFP and 116-LM/shYAP cells. (C) HCT116 cells were cotransfected with YAP5SA and GFP-PKM2 plasmids, and translocation of GFP-tagged PKM2 into the nucleus was detected by Western blot using an anti-GFP antibody. (D) Western blot analysis of PKM2 levels in 116-LM/shYAP cells in the presence of cycloheximide (CHX; 50 μg/ml). Quantification of PKM2 signals was performed using ImageJ. (E) Coimmunoprecipitation assay of the interaction between endogenous YAP and PKM2 in 116-LM cells (left panel). Cell lysates from HCT116 cells transfected with YAP5SA-flag and GFP-PKM2 plasmids were obtained, and coimmunoprecipitation analyses were carried out (right panel). (F) 116-LM cells were treated with gefitinib (10 μM) for 6 h, and cell lysates were immunoprecipitated with anti-YAP antibody, followed by immunoblotting with an anti-phospho-PKM2 antibody. The membrane was stripped and probed with an anti-PKM2 antibody. (G and H) Identification of the binding sites of PKM2 and YAP. Regions of interaction between PKM2 and YAP are indicated (G). Coimmunoprecipitation analyses of the deletion mutants of PKM2 and YAP5SA in HCT116 cells were carried out (H). Closed triangles denote the interaction of PKM2 with YAP, and open triangles denote the heavy chain and light chain. (I) HCT116 cells were transfected with YAP5SA and PKM2 plasmids, and the promoter activity of Glut3 was determined by luciferase reporter assay. (J) 116-LM cells were silenced with YAP, PKM2, or YAP/PKM2, and protein expression was analyzed by Western blot. (K) Capacities of tumor migration and invasion were determined by Transwell assays. *P<0.05, **P<0.01, ***P<0.001, determined by unpaired two-tailed Student's t-test. Quantitative analyses of all Western blots were carried out using ImageJ software.
To further study the mechanisms by which YAP regulates PKM2 in colorectal cancer, we examined the possibility that YAP interacts with PKM2. Interestingly, coimmunoprecipitation assays showed an interaction between endogenous YAP and PKM2 in 116-LM cells (Figure 4E left panel). These findings were confirmed by ectopic overexpression of YAP and PKM2 (Figure 4E right panel). Next, because the receptor tyrosine kinase signal regulates the phosphorylation of PKM2, we wanted to examine whether PKM2 phosphorylation promotes an interaction with YAP. 116-LM cells were treated with gefitinib, an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor that blocks PKM2 phosphorylation [14]. The results showed that although total PKM2 was unaffected by gefitinib treatment, the levels of YAP and phosphor-PKM2 simultaneously decreased (Figure 4F). Importantly, the interaction of YAP and PKM2 was impeded in the presence of gefitinib (Figure 4F), suggesting that the dysregulation of mitogen signaling in metastatic colorectal cancer induces PKM2 phosphorylation and consequently facilitates its association with YAP.
To further explore the molecular basis of the interaction between YAP and PKM2, we constructed a series of deletion mutants of PKM2 and YAP. The results of the coimmunoprecipitation assay revealed that the Δ1 mutant of PKM2, which lacked the B, A2, and C domains, did not bind YAP. However, the Δ3 mutant of PKM2, lacking the C domain, did bind to YAP, suggesting that the B and A2 domains of PKM2 are responsible for the association with YAP (Figure 4G and 4H). Moreover, we found that deletion of the TEAD binding domain in YAP (YAP-M2) abrogated the binding with PKM2 (Figure 4G and 4H).
To better understand whether PKM2 participates in the regulation of Glut3, a luciferase reporter assay was employed, and the results showed that overexpression of PKM2 and YAP5SA increased the promoter activity of Glut3 by 1.7- and 8-fold, respectively. Importantly, cotransfection with YAP5SA and PKM2 significantly increased the promoter activity of Glut3 compared with that of cells transfected with YAP or PKM2 alone (Figure 4I). Similar results were observed with Glut3 protein (Figure S6B). Moreover, knockdown of PKM2 not only suppressed Glut3 but also impeded YAP (Figure 4J), and compared with shYAP or shPKM2 treatments, the inhibition of both YAP and PKM2 substantially repressed Glut3 expression and tumor migration/invasion (Figure 4K). These data together indicate that oncogenic signaling facilitates the association of PKM2 with YAP and consequently promotes Glut3 expression and the invasiveness of CRC.
YAP plays a crucial role in metabolic changes in metastatic CRC
To perceive the impacts of YAP on metabolic changes in metastatic CRC, quantitative analysis of cellular metabolites in HCT116, 116-LM, and 116-LM/shYAP cells was performed using CE-TOF/MS. A total of 101 metabolites were identified (Figure S7A and S7B, and Table S2). We evaluated metabolic parameters using absolutely quantified data obtained from CE-TOF/MS to investigate the physiological states of the cells (Table S3). First, we found that the glucose 6-phosphate (G6P) to ribose 5-phosphate (R5P) ratio, a parameter that shows the significance of the activity of glycolysis and the pentose phosphate pathway, was increased over 2-fold in 116-LM cells compared with HCT116 cells (Figure 5A). The pentose phosphate pathway is a major source of purine metabolism. Our data showed that the levels of total adenylate and guanylate, which are related to de novo purine synthesis, were increased in 116-LM cells (Figure 5B). Likewise, 116-LM cells showed a higher glycerol 3-phosphate/dihydroxyacetone phosphate (DHAP) ratio, which is an indirect parameter reflecting the NADH/NAD+ ratio and energy status (Figure 5C). Indeed, ATP production was also higher in 116-LM cells (Figure 5B). In addition, amino acids, both essential and nonessential, were present at higher levels in 116-LM cells (Figure 5D). Specifically, the glucogenic amino acids, which are used in gluconeogenesis, were increased approximately 2-fold in 116-LM cells (Figure 5D). These data revealed that the synthesis of macromolecules was elevated in 116-LM cells and highlighted the importance of glucose utilization and energy demand in CRC metastasis.
Moreover, we found that reduced glutathione (GSH) was upregulated in 116-LM cells (Figure 5E). Glutathione removes and detoxifies carcinogens, which confers resistance to chemotherapeutic drugs and protects cancer cells. In contrast, the GSH to oxidized glutathione (GSSG) ratio, which reflects oxidative stress, was increased in YAP-knockdown 116-LM cells (Figure 5E). These data also revealed the indispensable role of YAP in drug resistance. However, these metabolic alterations were decreased in YAP-knockdown 116-LM cells (Figure 5A-E). Notably, the adenylate and guanylate energy charges were not affected in the three groups (Figure 5B). Energy charge is a cellular metabolic indicator that decreases under severe stress conditions. Together with the aforementioned metabolic parameters, these data demonstrated that YAP plays a crucial role in shaping metabolic reprogramming in CRC metastasis.
Figure 5
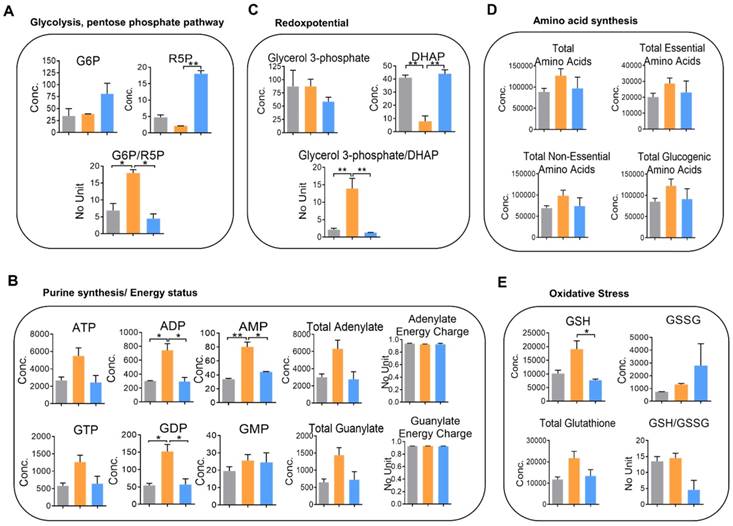
Knockdown of YAP alters cellular metabolism in 116-LM cells. (A-E) Absolute quantitative analysis of metabolites in HCT116 (gray color), 116-LM (yellow color) and 116-LM/shYAP (blue color) cells. The metabolic parameters for glycolysis/the pentose phosphate pathway (A), purine synthesis/energy status (B), redox potential (C), amino acid synthesis (D), and oxidative stress (E) are shown. The results are representative of two independent CE-TOF/MS experiments. Bar, SD. Concentration= pmole/106 cells. Statistical analysis was performed using ANOVA.
The Glut3/YAP axis is indispensable for high-fat/sucrose diet-mediated CRC metastasis and is a potential therapeutic target
To gain insights into the significance of high glucose-elicited tumor progression, we fed immunocompromised mice a diet containing 45% fat and 17% sucrose (HFSD), which reflects Western food-mediated metabolic changes and obesity in humans. Mice fed a HFSD showed an approximately 10% increase in body weight compared with mice receiving the standard chow diet (CD) (Figure 6A). However, fasting glucose was significantly elevated—by approximately 25%—in the HFSD-treated group (Figure 6B). Importantly, the metastatic burden was significantly increased in mice fed a HFSD compared with those fed a CD, whereas Glut3 suppression decreased the metastatic potential in both the CD- and HFSD-treated groups (Figure 6C). Silencing Glut3 had a greater impact on tumor metastasis in the HFSD group than in the CD group. These findings indicate that Glut3 indeed plays a crucial role in high sucrose-mediated CRC metastasis.
Since there is no treatment that currently targets Glut3, to further explore therapeutic strategies based on Glut3-YAP axis-mediated CRC metastasis, we investigated the potentiality of panobinostat, an FDA-approved histone deacetylase inhibitor (HDACi) whose inhibitory activity against colorectal and lung cancers we have previously identified by suppressing YAP and TAZ [12, 15]. As expected, panobinostat treatment not only decreased YAP protein levels but also suppressed Glut3 in three CRC lines (Figure 6D). Moreover, total and phosphorylated PKM2 were decreased by panobinostat (Figure 6D). The results of Transwell assays also showed that panobinostat effectively suppressed the invasiveness of colorectal cancer cells (Figure 6E). Thus, we further examined the potential of panobinostat to act on HFSD-associated tumor development. Immunocompromised mice were fed an HFSD, and the average body weights were 20% greater than those of the CD group (data not shown). The results consistently showed that mice receiving HFSD exhibited greater metastatic burden; however, treatment with panobinostat significantly reduced metastatic burden in both the CD and HFSD groups (Figure 6F). Accordingly, the fasting glucose concentration was increased in the HFSD treatment (Figure 6G), but no significant difference was found between the vehicle- and panobinostat-treated groups (Figure 6G), suggesting that the inhibitory effect of panobinostat was independent of blood glucose. These data indicate that targeting YAP-mediated metabolic reprogramming might be therapeutically beneficial in the treatment of CRC metastasis.
Figure 6
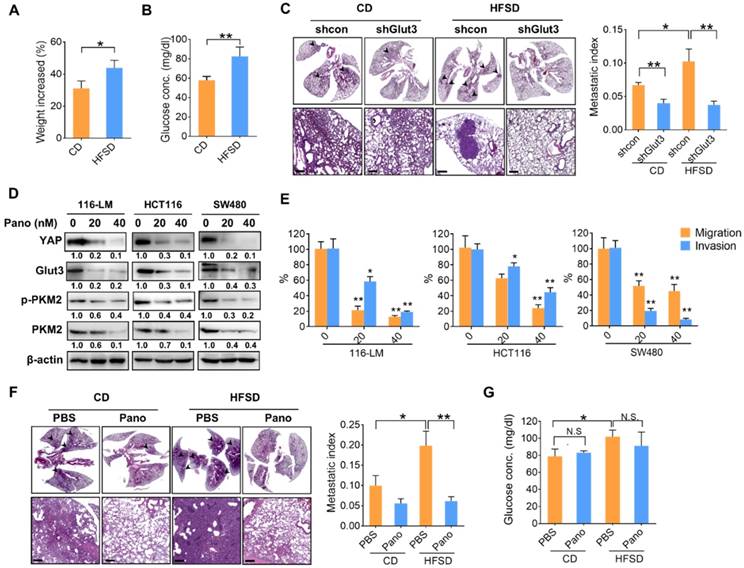
A high-fat sucrose diet promotes tumor metastasis in a YAP-Glut3-dependent signaling axis. (A and B) The increased body weight (A) and fasting glucose concentration (B) of BALB/c nude mice fed a high-fat/sucrose diet (HFSD) or standard chow diet (CD). N=5 in each group. (C) Mice fed a high-fat/sucrose diet (HFSD) showed increased metastatic colonization of 116-LM tumors, whereas knockdown of Glut3 abrogated metastatic colonization. Representative H&E images of mouse lung sections at low- (upper panel) and high-power (lower panel) magnifications are presented. Scale bar=200 μm. The metastatic index indicates the ratio of the area of metastatic burden to the total area of lung. (D and E) Colon cancer cells were treated with panobinostat (0, 20, 40 nM) for 24 h. The expression of glycolytic genes was analyzed by Western blot (D), and the capacity of tumor migration and invasion was evaluated by Transwell assay (E). Quantitative analyses of all Western blots were carried out using ImageJ software. (F and G) NOD/SCID mice were fed an HFSD or a CD for 7 weeks followed by intraperitoneal injection with panobinostat (8 mg/kg) for an additional 6 weeks. N=4 in each group. The metastatic burden was quantified as described above (F), and the fasting glucose concentration was analyzed (G). Representative H&E images of mouse lung sections at low- (upper panel) and high-power (lower panel) magnifications are presented. Scale bar=200 μm. The results are presented as the mean ± SEM. Significant differences were analyzed by ANOVA or t-test. N.S. denotes a nonspecific difference.
Discussion
Emerging evidence indicates that the Hippo transducer YAP/TAZ regulates metabolic reprogramming in cancer cells. Certain metabolic enzymes involved in glucose and amino acid metabolism are targets of YAP/TAZ. YAP transcriptionally regulates hexokinase 2 (HK2) and PFKFB3 [16], and YAP and TEAD1 work together to regulate Glut1 expression [17, 18]. Moreover, a recent study has shown that YAP promotes Glut3 transcription in glioma [13]; however, the detailed regulatory mechanism and its clinical relevance are unclear. In our study, we found that YAP and glycolytic genes were concomitantly elevated in metastatic CRC. We further identified the binding and transactivation of YAP on the Glut3 promoter region with a TEAD. Suppression of YAP downregulated the expression of glycolytic genes, including Glut3. Clinically, CRC patients show a highly positive correlation between YAP/TAZ and Glut3, and those whose tumors exhibit YAP and Glut3 coexpression have the poorest survival outcomes. Moreover, we showed that YAP plays an indispensable role in metabolic adaptation to metastatic colon cancer. Using metabolic parameters, we demonstrate that YAP regulates glycolysis, nucleotide synthesis, amino acid flux, and glutathione balance. Recently, Santinon and colleagues reported that the regulation of deoxynucleotide metabolism by YAP/TAZ is required for chemotherapeutic resistance [19]. We found that YAP regulates nucleotide biosynthesis and cellular detoxification; thus, chemotherapeutics may take advantage of metabolic alterations in cells requiring YAP. Together, the metabolic adaptation and the increased aerobic glycolysis caused by YAP may be part of a novel strategy for the treatment of colon cancer.
Recent findings have implicated YAP/TAZ activity in the regulation of metabolic cues, including glucose levels [20, 21]. In fact, glucose-induced upregulation of YAP/TAZ has been found not only by the inhibition of Hippo kinase via AMPK but also by interactions with the glycolytic enzyme PFK1 [20, 22]. Moreover, a recent study by Peng and colleagues showed that glucose-induced YAP glycosylation and activation promoted tumorigenesis [23]. In our study, we found that the upregulation of YAP in 116-LM cells occurs at least partially through Glut3, which facilitates glucose uptake and therefore increases YAP/TAZ. Suppression of Glut3 significantly diminished YAP and its downstream targets. Because YAP/TAZ plays a pivotal role in tumor progression, we also found that YAP regulates the expression of glycolytic genes. Our data indicate that positive feedback by Glut3 and YAP is present in the promotion of glucose metabolism and tumor metastasis.
Increasing evidence has revealed the crucial role of oncogenic PKM2 in tumor growth and stemness, independent of its pyruvate kinase activity. Phosphorylation of PKM2 causes it to translocate to the nucleus and function as a transcriptional cofactor of HIF-1 to stimulate the expression of glycolytic genes, including LDHA, PDK1, and SLC2A1 (Glut1) [24]. In addition, nuclear PKM2 associates with the oncoprotein c-Myc to regulate cell proliferation and metabolic reprogramming. Moreover, a recent study by Zhou and colleagues showed that PKM2 phosphorylation regulates YAP activity and thereby promotes the transformation of breast epithelial cells [25]; however, the detailed regulatory mechanism is unclear. Herein, we found that metastatic CRC upregulates the expression of total PKM2 and phospho-PKM2, which facilitates the association with YAP. YAP stabilizes and promotes nuclear translocation of PKM2, thereby enhancing Glut3 expression. Interestingly, we also found that PKM2 silencing affected YAP expression. These data are concordant with previous findings and suggest that PKM2 and YAP may interplay to each other. PKM2 has been reported to interact with the nuclear transporter Pin1 by binding the WW domain of Pin1 [7]. Our data showed that the TEAD binding domain rather than the WW domain in YAP is responsible for its association with PKM2. Given that the TEAD-YAP complexes predominantly govern YAP downstream gene expression, nuclear PKM2 plays a crucial role in shaping YAP-promoted tumorigenesis. Intriguingly, PKM2 suppression drastically impeded Glut3 expression, suggesting that other factors such as HIF-1 may cooperate with PKM2 to regulate the expression of Glut3. Further study is necessary to determine the possible mechanism of PKM2 phosphorylation and the genetic regulation of cancer metabolism by YAP/PKM2.
Conclusion: Our study indicated that Glut3 is an independent tumor marker for metastatic CRC. We identified the role of Glut3 in the promotion of CRC metastasis through a positive regulatory loop with YAP and PKM2 and suggest that it is a potential therapeutic target. The results of our study highlights that metabolic reprogramming is a feasible entry point for the treatment of colon cancer.
Materials and Methods
Cell culture
Human colorectal cancer cell lines were purchased from the Bioresource Collection and Research Center (Hsinchu, Taiwan). 116-LM cells were obtained from lung metastatic tumor cells by tail-vein injection of HCT116 cells into nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice, as established previously [12]. HCT15 and HT29 cells were cultured in RPMI. HCT116, 116-LM, SW480, and SW620 cells were cultured in DMEM. All culture media were supplemented with 7% fetal bovine serum and GlutaMAX (Gibco). All cell lines were authenticated through short tandem repeat profiling (GenePrint 10 System) to ensure that no culture contamination had occurred.
Reagents and plasmids
2-Deoxy-glucose (2DG) was purchased from Sigma. Panobinostat was obtained from Selleckchem. The 8xTGIIC luciferase reporter plasmid was provided by Dr. Stefano Piccolo (Addgene #34615). The pcDNA-flag-YAP plasmid was provided by Dr. Marius Sudol (Addgene #17793). The pCMV-flag-YAP5SA plasmid was provided by Dr. Kunliang Guan (Addgene #27371). The pEGFP-C1-PKM2 plasmid was provided by Dr. Axel Ullrich (Addgene #64698). Full-length human Glut3 in the pcDNA3/DYK vector was purchased from GenScript. The deletion mutants of YAP5SA and PKM2 were subcloned into the pcDNA3-HA vector. The promoter of Glut3 (-2100~+43) was constructed in the pGL4.1[luc2] reporter plasmid. The promoters of Oct4, Nanog, and c-Myc were previously constructed in pGL4.1[luc2] reporter plasmids [26].
Expression of the Glut family in human colon cancer patients
Gene expression patterns of the Glut family and the clinicopathological profile of human CRC were obtained from TCGA, Hong [27], and Skrzypczak [28] datasets (Oncomine). The survival prognosis of different cancer types as a function of Glut3 was determined using OncoLnc and PrognoScan databases. A high expression group was defined as greater than 30% of patients. The RNA expression value of 7 paired primary and liver metastatic colon cancer tissues was obtained from the GSE40367 dataset [29]. Microarray data were analyzed with R version 3.3.1 software. GSEA was performed using the MSigDB. The correlation of Glut3 and the YAP signature (CORDENONSI_YAP_CONSERVED_SIGNATURE) in CRC patients were obtained from GSE41258 [30], GSE68468, GSE40967 [31], and GSE17536 [32] datasets.
Measurement of glycolytic capacity
The real-time extracellular acidification rate (ECAR) was measured using a Seahorse XFe24 Flux Analyzer (Seahorse Bioscience) according to the manufacturer's instructions. Cells were seeded in a Seahorse plate overnight to reach 80% confluence. One hour before measurement, the culture medium was replaced with XF basal medium (Seahorse Bioscience). The ECAR was measured over time following the injection of 10 mM glucose, 10 μM oligomycin, and 50 mM 2-deoxyglucose (2-DG). Glycolytic capacity was normalized to the cell number.
Metabolite measurements
A total of 1x107 cells were washed with ice-cold phosphate-buffered saline (PBS), and their metabolism was quenched with 500 μl of methanol. Then, the sample was vortexed for 2 min and kept on ice for 5 min. Another 500 μl of cold water was added, and the treatment was repeated. The supernatant was collected by centrifuging the sample at 15,000 rcf for 5 min, and 100 μl of the extracted supernatant was dried with a SpeedVac. The dried residues were further derivatized with trimethylsilylation, and the samples were analyzed by a gas chromatography-mass spectrometer (GC-MS) (Agilent 7890A) coupled with a 5975C inert mass selective detector (MSD) for metabolic profiling. Heatmaps were generated by MetaboAnalyst 4.0.
Targeted quantitative analysis of cellular metabolites was conducted by the C-SCOPE package of HMT (Human Metabolome Technologies, Inc., HMT) using capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS) for cation analysis and CE-tandem mass spectrometry (CE-MS/MS) for anion analysis. Briefly, 3-5x106 cells were washed twice with 5% mannitol solution. The cells were then treated with methanol and then treated with Milli-Q water containing internal standards (H3304-1002, Human Metabolome Technologies (HMT), Tsuruoka, Yamagata, Japan). The extract was filtered through a Millipore 5-kDa cutoff filter (UltrafreeMC-PLHCC, HMT) to remove macromolecules. The filtrate was centrifugally concentrated and resuspended in Milli-Q water for metabolomic analysis at HMT. The metabolites were annotated based on the HMT metabolite database. Hierarchical cluster analysis (HCA) and principal component analysis (PCA) were performed by statistical analysis software (Human Metabolome Technologies, Inc.). The metabolic parameters were evaluated using the equation described in Table S3. All values were normalized to cell numbers.
Short hairpin RNA and lentiviral infection
Short hairpin RNAs (shRNAs) for human YAP (TRCN0000107265; TRCN0000107266) and PKM2 (TRCN0000195581; TRCN0000195588) were obtained from the National RNAi Core Facility (Academia Sinica, Taipei, Taiwan). Lentiviral preparation and virus infection were performed as previously described. In brief, HEK293T cells were cotransfected with pLKO.shRNA together with pCMV-∆R8.91 and pMDG plasmids. At 48-h posttransfection, virus-containing supernatants were incubated with target cells for another 48 h. Transduced cells were selected using puromycin (2 μg/ml). A control shRNA targeting red fluorescent protein (RFP) was used as a negative control. A pool of Glut3 lentiviral particles containing 3 target-specific shRNAs (sc-41218V) was obtained from Santa Cruz Biotechnology. For CRISPR/Cas9-mediated Glut3 knockout, 116LM/sgGlut3 cells were generated using the All-in-one sgRNA/Cas9 system (National RNAi core, Academia Sinica, Taipei, Taiwan). The shRNA sequences are shown in Table S4.
Cell migration and invasion assays
For cell migration, 1x105 cells in 200 μl of serum-free medium were seeded into the upper chamber of a Transwell insert. For cell invasion, 4x105 cells in 200 μl of serum-free medium were seeded into the upper chamber of a Matrigel (BD Biosciences)-coated Transwell insert. The lower wells were filled with complete medium and served as the chemo-attractant. After 24 h, the cells were fixed with methanol and stained with crystal violet. The Matrigel and unmigrated cells were removed using cotton swabs prior to analysis.
Tumorsphere formation assay
Cells (104) were seeded in six-well ultralow attachment plates (Corning Costar) and maintained in serum-free DMEM/F12 supplemented with 20 ng/ml epidermal growth factor (PeproTech), 25 ng/ml basic fibroblast growth factor (PeproTech), and B27 (Gibco) for 10 days. The formation of tumorspheres was observed, and they were counted under a light microscope.
Immunoprecipitation and Western blotting
Cells were lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer supplemented with a protease and phosphatase inhibitor cocktail (Roche). Whole-cell extracts were incubated with the indicated antibodies and protein A/G beads (Santa Cruz Biotechnology) overnight. The beads were washed with RIPA buffer three times and then boiled in SDS loading buffer. For Western blotting, cell extracts were separated by SDS-PAGE. After Western transfer, membranes were probed with antibodies against DDK (Origene ta50011), GFP (Santa Cruz biotechnology sc-101536), Glut3 (Santa Cruz biotechnology sc-74399), HA (Santa Cruz biotechnology sc-393579), PKM2 (Cell Signaling #3198), phospho-PKM2(Y705) (Cell Signaling #3827), YAP (Cell Signaling #12395), phospho-YAP(S127) (Cell Signaling #13008), actin (GeneTex GTX11003) and lamin C (GeneTex GTX101127). Bands were detected with an enhanced chemiluminescence system (Millipore). Western blotting was performed at least three times, and representative experiments are shown. Quantification was carried out using ImageJ software.
Luciferase reporter assay
Cells were seeded in 24-well plates and transiently transfected with a pGL4.1[luc2]-promoter reporter plasmid together with the Renilla luciferase (RL)-TK plasmid using PolyJET transfection reagent. Firefly and Renilla luciferase activities were measured using a Dual-Luciferase Assay System (Promega) according to the manufacturer's instructions.
Real-time PCR
Total RNA was extracted with an RNA extraction kit and reverse-transcribed with a high-capacity cDNA conversion kit (Invitrogen, Carlsbad, CA, USA). cDNA was amplified using EvaGreen Master Mix (Biotium, Hayward, CA, USA) using a StepOne Plus Real-Time PCR system (Applied Biosystems, Darmstadt, Germany) with specific primers. The results were calculated using the ΔΔCT equation and are expressed as multiples of change relative to a control sample.
Chromatin immunoprecipitation
The protein-DNA complexes were crosslinked using 1% formaldehyde, which was then quenched by adding glycine to a final concentration of 200 mM. Chromatin complexes were sonicated to an average size of 250 bp by a MISONIX Sonicator 3000. The chromatin was incubated with anti-YAP (Cell Signaling #14074) and protein A/G beads (EZ-Magna ChIP Kit, Millipore) overnight. The immunocomplexes were reverse crosslinked, and the purified DNA was subjected to PCR analysis. The PCR primers are listed in Table S5.
Immunohistochemistry
Colorectal cancer tissue microarrays were purchased from Biomax. Slides were deparaffinized, rehydrated and heated in an antigen-unmasking solution (Vector Laboratories). Slides were incubated first with anti-Glut3 primary antibody (Santa Cruz biotechnology) overnight at 4 °C and then with SignalStain Boost IHC detection reagent (Cell Signaling Technology), and they were then visualized by 3,3'-diaminobenzidine (DAB) peroxidase substrate (Vector Laboratories). The H-score was determined by estimating the percentage of tumor cells positively stained with weak, moderate, and strong intensity and calculation using the following formula: (low%) × 1 + (medium%) × 2 + (high%) × 3.
Animal study
All animal studies were performed according to the guidelines and approval of the Animal Care and Use Committee of Taipei Medical University. To study the effect of Glut3 on CRC metastasis, 5-week-old BALB/c nude mice were fed a diet containing 45% fat and 17% sucrose (high-fat/sucrose diet (HFSD)) (Research Diets) or a standard chow diet (CD). Meanwhile, mice were injected intravenously with 2x106 116-LM/shRFP and 116-LM/shGlut3 cells for 7 weeks. To study the effect of panobinostat on colorectal cancer metastasis, 5-week-old NOD/SCID mice were fed an HFSD or CD for 7 weeks, followed by intravenous injection with 116-LM cells. After one week of inoculation with tumor cells, mice were given an intraperitoneal injection with vehicle or panobinostat (8 mg/kg, every T and W, weekly) for an additional 6 weeks. All dissected lungs were embedded with paraffin, and tissue sections were stained with hematoxylin and eosin and scanned by TissueFax (TissueGnostics). The metastatic index was calculated as the ratio of the area of metastatic burden to the total area of lung. To measure fasting plasma glucose levels, mice were fasted for 6 or 9 h during the day. Food was removed, but mice were allowed free access to water. Blood glucose levels were measured using a blood glucometer (Roche Diagnostics).
Statistical analyses
Data are presented as the mean ± standard error (SE) of three independent experiments. Statistical significance was determined by an unpaired, two-tailed Student's t-test unless stated otherwise. * Indicates a p value <0.05; ** indicates a p value <0.01. Coexpression trends were analyzed by a chi-squared test. A correlation coefficient was analyzed by the Pearson test. The survival probability was analyzed by a log-rank (Mantel-Cox) statistical test. Statistical analysis was carried out using GraphPad Prism software.
Abbreviations
2DG, 2-deoxyglucose; CE-TOF/MS, capillary electrophoresis time-of-flight mass spectrometry; ChIP, chromatin immunoprecipitation; CRC, colorectal cancer; ECAR, extracellular acidification rate; EMT, epithelial-mesenchymal transition; HFSD, high-fat/sucrose diet; GC, gas chromatography; Glut3, glucose transporter 3; GSEA, gene set enrichment algorithm; TCGA, The Cancer Genome Atlas; PKM2, pyruvate kinase M2; YAP, Yes-associated protein.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by grants from the Ministry of Science and Technology, Taiwan (MOST106-2320-B-038-040 and MOST107-2320-B-038-050-MY3), and the Chi Mei Medical Center (106CM-TMU-10 and 107CM-TMU-07). We thank Metabolomics Core Laboratory at the Centers of Genomic and Precision Medicine of National Taiwan University for performing the metabolomics experiments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. van der Geest LG, Lam-Boer J, Koopman M, Verhoef C, Elferink MA, de Wilt JH. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clin Exp Metastasis. 2015;32:457-65
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
3. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441-64
4. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M. et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797-800
5. Liang J, Cao R, Zhang Y, Xia Y, Zheng Y, Li X. et al. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat Commun. 2016;7:12431
6. Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci U S A. 2012;109:E3367-76
7. Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F. et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14:1295-304
8. Thorens B, Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. 2010;298:E141-5
9. Crippa S, Ancey PB, Vazquez J, Angelino P, Rougemont AL, Guettier C. et al. Mutant CTNNB1 and histological heterogeneity define metabolic subtypes of hepatoblastoma. EMBO Mol Med. 2017;9:1589-604
10. Masin M, Vazquez J, Rossi S, Groeneveld S, Samson N, Schwalie PC. et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. 2014;2:11
11. Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE. et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16:1373-82
12. Ling HH, Kuo CC, Lin BX, Huang YH, Lin CW. Elevation of YAP promotes the epithelial-mesenchymal transition and tumor aggressiveness in colorectal cancer. Exp Cell Res. 2017;350:218-25
13. Cosset E, Ilmjarv S, Dutoit V, Elliott K, von Schalscha T, Camargo MF. et al. Glut3 Addiction Is a Druggable Vulnerability for a Molecularly Defined Subpopulation of Glioblastoma. Cancer Cell. 2017;32:856-68 e5
14. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W. et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118-22
15. Lee WY, Chen PC, Wu WS, Wu HC, Lan CH, Huang YH. et al. Panobinostat sensitizes KRAS-mutant non-small-cell lung cancer to gefitinib by targeting TAZ. Int J Cancer. 2017;141:1921-31
16. Zheng X, Han H, Liu GP, Ma YX, Pan RL, Sang LJ. et al. LncRNA wires up Hippo and Hedgehog signaling to reprogramme glucose metabolism. EMBO J. 2017;36:3325-35
17. Lin C, Xu X. YAP1-TEAD1-Glut1 axis dictates the oncogenic phenotypes of breast cancer cells by modulating glycolysis. Biomed Pharmacother. 2017;95:789-94
18. Valis K, Talacko P, Grobarova V, Cerny J, Novak P. Shikonin regulates C-MYC and GLUT1 expression through the MST1-YAP1-TEAD1 axis. Exp Cell Res. 2016;349:273-81
19. Santinon G, Brian I, Pocaterra A, Romani P, Franzolin E, Rampazzo C. et al. dNTP metabolism links mechanical cues and YAP/TAZ to cell growth and oncogene-induced senescence. EMBO J. 2018:37
20. Enzo E, Santinon G, Pocaterra A, Aragona M, Bresolin S, Forcato M. et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015;34:1349-70
21. Koo JH, Guan KL. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018;28:196-206
22. Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL. et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015;17:490-9
23. Peng C, Zhu Y, Zhang W, Liao Q, Chen Y, Zhao X. et al. Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol Cell. 2017;68:591-604 e5
24. Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin YS, Yang SF. et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1alpha-mediated glucose metabolism. Proc Natl Acad Sci U S A. 2014;111:279-84
25. Zhou Z, Li M, Zhang L, Zhao H, Sahin O, Chen J. et al. Oncogenic Kinase-Induced PKM2 Tyrosine 105 Phosphorylation Converts Nononcogenic PKM2 to a Tumor Promoter and Induces Cancer Stem-like Cells. Cancer Res. 2018;78:2248-61
26. Lin CW, Liao MY, Lin WW, Wang YP, Lu TY, Wu HC. Epithelial cell adhesion molecule regulates tumor initiation and tumorigenesis via activating reprogramming factors and epithelial-mesenchymal transition gene expression in colon cancer. J Biol Chem. 2012;287:39449-59
27. Edwards S, Campbell C, Flohr P, Shipley J, Giddings I, Te-Poele R. et al. Expression analysis onto microarrays of randomly selected cDNA clones highlights HOXB13 as a marker of human prostate cancer. Br J Cancer. 2005;92:376-81
28. Skrzypczak M, Goryca K, Rubel T, Paziewska A, Mikula M, Jarosz D. et al. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One. 2010:5
29. Roessler S, Lin G, Forgues M, Budhu A, Hoover S, Simpson RM. et al. Integrative genomic and transcriptomic characterization of matched primary and metastatic liver and colorectal carcinoma. Int J Biol Sci. 2015;11:88-98
30. Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F. et al. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc Natl Acad Sci U S A. 2009;106:7131-6
31. Marisa L, de Reynies A, Duval A, Selves J, Gaub MP, Vescovo L. et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453
32. Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A. et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology. 2010;138:958-68
Author contact
Corresponding author: Cheng-Wei Lin; Department of Biochemistry and Molecular Biology, School of Medicine, College of Medicine, Taipei Medical University, 250 Wu-Xing Street, Taipei 110, Taiwan Email: cwlinedu.tw; Phone: 886-2-27361661 ext 3160; Fax: 886-2-27356689
Received 2019-1-8
Accepted 2019-3-8
Published 2019-4-13
Citation styles
APA
Kuo, C.C., Ling, H.H., Chiang, M.C., Chung, C.H., Lee, W.Y., Chu, C.Y., Wu, Y.C., Chen, C.H., Lai, Y.W., Tsai, I.L., Cheng, C.H., Lin, C.W. (2019). Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit. Theranostics, 9(9), 2526-2540. https://doi.org/10.7150/thno.32915.
ACS
Kuo, C.C.; Ling, H.H.; Chiang, M.C.; Chung, C.H.; Lee, W.Y.; Chu, C.Y.; Wu, Y.C.; Chen, C.H.; Lai, Y.W.; Tsai, I.L.; Cheng, C.H.; Lin, C.W. Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit. Theranostics 2019, 9 (9), 2526-2540. DOI: 10.7150/thno.32915.
NLM
Kuo CC, Ling HH, Chiang MC, Chung CH, Lee WY, Chu CY, Wu YC, Chen CH, Lai YW, Tsai IL, Cheng CH, Lin CW. Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit. Theranostics 2019; 9(9):2526-2540. doi:10.7150/thno.32915. https://www.thno.org/v09p2526.htm
CSE
Kuo CC, Ling HH, Chiang MC, Chung CH, Lee WY, Chu CY, Wu YC, Chen CH, Lai YW, Tsai IL, Cheng CH, Lin CW. 2019. Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit. Theranostics. 9(9):2526-2540.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.