ISSN: 1838-7640Theranostics
- Current issue
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Volume 11; 2021
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Top
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
Global reach, higher impact
Theranostics 2017; 7(5):1088-1099. doi:10.7150/thno.18551 This issue Cite
Research Paper
Exome Sequencing of Oral Squamous Cell Carcinoma Reveals Molecular Subgroups and Novel Therapeutic Opportunities
Shih-Chi Su1,2, Chiao-Wen Lin3,4, Yu-Fan Liu5, Wen-Lang Fan1, Mu-Kuan Chen6, Chun-Ping Yu7, Wei-En Yang8,9, Chun-Wen Su8,9, Chun-Yi Chuang10,11, Wen-Hsiung Li7, Wen-Hung Chung1,2,12, Shun-Fa Yang8,9,
1. Whole-Genome Research Core Laboratory of Human Diseases, Chang Gung Memorial Hospital, Keelung, Taiwan;
2. Department of Dermatology, Drug Hypersensitivity Clinical and Research Center, Chang Gung Memorial Hospital, Linkou, Taiwan;
3. Institute of Oral Sciences, Chung Shan Medical University, Taichung, Taiwan;
4. Department of Dentistry, Chung Shan Medical University Hospital, Taichung, Taiwan;
5. Department of Biomedical Sciences, Chung Shan Medical University, Taichung, Taiwan;
6. Department of Otorhinolaryngology-Head and Neck Surgery, Changhua Christian Hospital, Changhua, Taiwan;
7. Biodiversity Research Center, Academia Sinica, Taipei, Taiwan;
8. Institute of Medicine, Chung Shan Medical University, Taichung, Taiwan;
9. Department of Medical Research, Chung Shan Medical University Hospital, Taichung, Taiwan;
10. School of Medicine, Chung Shan Medical University, Taichung, Taiwan;
11. Department of Otolaryngology, Chung Shan Medical University Hospital, Taichung, Taiwan;
12. School of Medicine, College of Medicine, Chang Gung University, Taoyuan, Taiwan.
Citation:
Su SC, Lin CW, Liu YF, Fan WL, Chen MK, Yu CP, Yang WE, Su CW, Chuang CY, Li WH, Chung WH, Yang SF. Exome Sequencing of Oral Squamous Cell Carcinoma Reveals Molecular Subgroups and Novel Therapeutic Opportunities. Theranostics 2017; 7(5):1088-1099. doi:10.7150/thno.18551. https://www.thno.org/v07p1088.htm
Other stylesAbstract
Oral squamous cell carcinoma (OSCC), an epithelial malignancy affecting a variety of subsites in the oral cavity, is prevalent in Asia. The survival rate of OSCC patients has not improved over the past decades due to its heterogeneous etiology, genetic aberrations, and treatment outcomes. Improvement in therapeutic strategies and tailored treatment options is an unmet need. To unveil the mutational spectrum, whole-exome sequencing of 120 OSCC from male individuals in Taiwan was conducted. Analyzing the contributions of the five mutational signatures extracted from the dataset of somatic variations identified four groups of tumors that were significantly associated with demographic and clinical features. In addition, known (TP53, FAT1, EPHA2, CDKN2A, NOTCH1, CASP8, HRAS, RASA1, and PIK3CA) and novel (CHUK and ELAVL1) genes that were significantly and frequently mutated in OSCC were discovered. Further analyses of gene alteration status with clinical parameters revealed that the tumors of the tongue were enriched with copy-number alterations in several gene clusters containing CCND1 and MAP4K2. Through defining the catalog of targetable genomic alterations, 58% of the tumors were found to carry at least one aberrant event potentially targeted by US Food and Drug Administration (FDA)-approved agents. Strikingly, if targeting the p53-cell cycle pathway (TP53 and CCND1) by the drugs studied in phase I-III clinical trials, those possibly actionable tumors are predominantly located in the tongue, suggesting a better prediction of sensitivity to current targeted therapies. Our work revealed molecular OSCC subgroups that reflect etiological and prognostic correlation as well as defined the landscape of major altered events in the coding regions of OSCC genomes. These findings provide clues for the design of clinical trials for targeted therapies and stratification of OSCC patients with differential therapeutic efficacy.
Keywords: Oral squamous cell carcinoma, exome sequencing, mutational signature, driver gene, targeted therapy.
Introduction
Oral squamous cell carcinoma (OSCC) is the most common malignant disease developing in the oral cavity, accounting for the vast majority (approximately 90%) of oral cancers [1]. Despite current advances in surgery and other treatment options, the fatality of oral cancer has remained mostly unchanged in years [2]. The prevalence of oral cancers is high globally, especially in South and Southeast Asia [3]. Strikingly, the trend for OSCC has increased five-fold in men and doubled among women in Taiwan over the past decades [4]. Such variation in the distribution and incidence rate of this disease results from the combination of distinct risk factors associated with the unique cultural practice and life style of different ethnic populations. Major risk factors of oral cancer, including human papillomavirus (HPV) infection and habitual exposure of cancer-causing substances, such as tobacco and alcohol use and betel nut chewing have been well demonstrated [5]. In Taiwan, betel nut chewing is popular and predominantly correlated with an increased risk of OSCC [4], while the prevalence of oncogenic HPV infection showed a declining trend [6].
Besides, oral carcinogenesis is known to be modulated by genetic alterations that affect cell cycle, apoptosis, and DNA repair [7]. Previous exome-sequencing studies on head and neck squamous cell carcinoma (HNSCC) have consistently revealed that TP53, CDKN2A, PIK3CA, HRAS and NOTCH1 were significantly mutated [8, 9]. Another genomic analysis of OSCC unraveled four major driver pathways (mitogenic signalling, Notch, cell cycle, and TP53) and defined a novel molecular subtype of OSCC as characterized by frequent CASP8 mutations accompanied with few copy-number changes [10]. In addition, an investigation on the mutational landscape of OSCC at the gingivo-buccal region showed the identification of site-specific genes significantly and frequently altered (USP9X, MLL4, ARID2, UNC13C and TRPM3) in oral cancer [11]. Moreover, The Cancer Genome Atlas (TCGA) network has comprehensively characterized HPV- and HPV+ HNSCC, providing a foundation for advanced molecular diagnoses, identification of potential biomarkers, and therapeutic insights [12]. The collective somatic mutations within a cancer genome were elucidated to be the readout of certain oncogenic patterns that are associated with mutagenic exposures or defects in DNA maintenance [13-15]. Since OSCC etiology varies across geographical regions, it is conceivable that the outcome of mutagenic processes operative in OSCC may be unique in Asian countries.
To define the genomic landscape of oral cancer in Taiwan where both the age-standardized incidence for males and the ratio of male to female are among the highest in Asia [3], we characterized the mutational catalog of OSCC and explored their clinical relevance in Taiwanese male populations. Our study discovered the major druggable aberrations in OSCC genomes and provided potential avenues for a genome-based classification of OSCC patients with prognostic and therapeutic implications.
Materials and Methods
Collection of samples and clinical data
Tumors and paired whole blood samples of 136 male patients who had been neither previously treated nor proven metastatic disease at the time of diagnosis were collected in this study, with the approval by the institutional review board of Chung Shan Medical University Hospital, Taichung, Taiwan. Subjects were accrued from 2008 to 2015. Cases with enriched tumor cell populations to ensure the selection of regions > 70% tumor purity were selected by certified pathologists. All tumor samples were immediately frozen in liquid nitrogen and stored at -80 °C. All cases were staged clinically at the time of diagnosis according to the TNM staging system of the American Joint Committee on Cancer (AJCC) [16]. Tumor differentiation was examined by a pathologist and rated according to the AJCC classification. All participants provided informed written consent at enrollment. Data on age, gender, alcohol drinking, betel quid chewing, and cigarette smoking were recorded for each participant. Betel quid chewing and alcohol drinking are defined as behavioral use of betel nuts (or related products) and substantial alcohol intake, respectively. Cigarette smoking is defined as current smoking of at least one cigarette per day during the latest three months.
Sequencing
Genomic DNA was extracted by using the DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer's instructions and loaded on a 1% agarose gel for quality control. For whole-exome sequencing (WES), in-solution enrichment of coding exons and flanking intronic sequences for 120 pairs of qualified DNA samples was performed by the SureSelect Human All Exon V5 kit (Agilent Technologies). Library construction and sequencing on an Illumina HiSeq 2500 instrument with paired reads of 90-100 bp were performed by Novogene. For whole-genome sequencing (WGS), DNA libraries with an insert size of 500-600 bp were prepared according to the protocol provided by Illumina and sequenced on a set of Illumina HiSeq X instruments with paired reads of 150 bp.
Analysis of mutational signatures
We deciphered the mutational signatures by using the Wellcome Trust Sanger Institute mutational signatures framework [17]. This algorithm used a non-negative matrix factorization (NMF) method to express each signature as a matrix that is derived from all mutation data according to 96 possible mutation types (6 types of substitution * 4 types of 5' base * 4 types of 3' base). The optimal number of mutational signatures was identified based on reproducibility of signatures and low error for reconstructing the original catalogs by running permutation tests with increasing the number of signature from one to ten (Fig. S1). The identified signatures were compared to previously-extracted consensus signatures [13] by cosine similarity (Table S1).
Identification and verification of somatic mutations
Cleaned sequence data were aligned and mapped to the reference genome (hg19) by Burrows-Wheeler aligner (BWA) using default options [18]. We applied MuTect [19] and Strelka [20] to identify somatic single-nucleotide variations (SNVs) and small insertions and deletion (INDELs), respectively, in target regions plus 5' and 3' flanking fragment of 20 bp. All somatic mutations detected by whole-exome sequencing were compared to variant calls from a panel of 267 germline DNA samples (Chang Gung Human Database, an unpublished whole-genome database of normal controls). Non-recurrent mutations found in 2% of the germline samples were removed from analysis. A total of 143 mutations in TP53, CASP8, EPHA2, ELAVL1, CHUK, ASXL1, RPTN, and CHRNB4 were evaluated using Sanger sequencing (Table S2), and 135 were validated at a rate of 94.4%. In addition, a total of 951 (5.1%) putative somatic mutations were inspected manually using the Integrated Genomics Viewer (IGV).
Detection of significantly mutated genes
To identify significantly mutated genes, we applied MutSigCV [21], which corrects the presence of mutations with nucleotide context, gene expression, replication time, and observed silent mutations, with default options on sequence data including flanking fragments of 20 bp. Furthermore, IntOGen [22], which determines whether genes are enriched for possibly impactful variants beyond what is expected by chance, was run on an online package. An additional χ2-based algorithm as described [23] was used to recognize a bias toward inactivating mutations. Genes with a false discovery rate-corrected q value of <0.1 were considered significantly mutated.
Analysis of copy-number alterations
To detect somatic copy-number alterations (CNAs), we applied Control-FREEC [24] to the read count profiles of sequence data. The read count ratios of tumors to paired normal samples were calculated, normalized for GC-content and mappability, and used as the proxy of the copy-number ratios. We used GISTIC2.0 [25] to infer significantly recurrent CNAs. A G-score that evaluates the frequency and amplitude of the aberration across samples was assigned for each alteration. False discovery rate (FDR) q values were then computed for the aberrant regions. Peak regions with q values of <0.25 were considered significant. In addition, a residual q value, that is, the q value of a peak region after excluding events that overlap with other significant peak regions on the same chromosome was also assigned. Quantitative PCR was used for validating representative somatic CNAs detected in GISTIC analysis. All PCR reactions using TaqMan assays (Invitrogen) (Table S2) were performed in triplicate for each blood and tumor DNA samples with an Applied Biosystems 7900HT. Copy-number change of somatic CNAs in tumor with respect to blood was measured as previously described [26].
Analysis of recurrently targetable genes and pathways
Pathway-enrichment analyses of genes were performed with Gene Ontology by annotating the 35 candidate genes and defining their primary roles through an extensive review of pathway databases including GeneCards and KEGG, such that four major pathways altered in more than 10% of OSCC were identified. Relationships between genes and distinctions of the pathways in the subcellular compartments were depicted as presented in the KEGG pathway database. The FDA-approved agents or drugs tested in clinical trials that were related to our candidate genes were reviewed in the ClinicalTrails.gov and the Drug Dictionary databases from National Cancer Institute.
Statistical analyses
Student's t test was used to evaluate the significant differences in number of mutations between groups. The significance of association between the clusters and clinical data was tested using Fisher's exact test. The strength of association or exclusion among gene alterations was assessed by a binary logistic regression analysis. Other tests of independence were performed using the χ2 test, unless otherwise indicated. Data were analyzed by using the SAS statistical software (Version 9.1, 2005; SAS Institute Inc., Cary, NC). All reported p values were two-tailed, and a p value of <0.05 was considered significant.
Other Materials and Methods, including immunohistochemistry, immunoblotting, silencing of CHUK in OSCC cells, cell migration and invasion assay, and detection of somatic structural variants, are described in Supplementary Information.
Results
Clinical characteristics and genomic analyses of OSCC
To characterize the mutational spectrum, we conducted whole-exome sequencing of 120 OSCC tumor-normal (fresh-frozen tumors and matched whole blood) pairs, 2 of which have also been subjected to whole-genome sequencing. All patients recruited were male, and the average age at disease onset was 56 (Table S3). Various anatomical sites were represented, including buccal mucosa (40%), tongue (26.7%), lip (10%), gingiva (9.2%), and others (14.1%). Of the patients profiled in this study, 89.2%, 50%, and 79.2% reported a history of tobacco, alcohol, and betel nut use, respectively. Surgical dissection and histological examination confirmed advanced stage III/IV in 47.5% cases. Lymph node metastasis occurred in 30% of patients.
We conducted exome sequencing to a mean depth of 83.2- and 84.5-fold, with 89.2% and 89.4% of targeted regions covered at >20-fold in tumors and matched normal samples, respectively (Fig. S2). Coding regions of 120 OSCC genomes contained 18,501 somatic variants, of which 17771 (96.05%) were single-nucleotide variations (SNVs) and the remaining were small insertions and deletions (INDELs) (Table S4-6). Of these variants, a median of 69.5 silent and 59.5 non-silent mutations was detected per tumor (ranging from 3 to 1146), corresponding to a mean somatic mutation rate of 2.62 per megabase in coding sequencing, compatible with that of previous studies [8, 9, 11]. A higher number of somatic mutations were observed in large-sized tumors and in elder patients (Fig. S3).
Mutational signatures in OSCC
To define the mutational signatures operative in OSCC, we applied non-negative matrix factorization (NMF) [17] and deciphered five mutational signatures in our cohort (Fig. 1A, Table S1 and Fig. S1). Among these, four signatures that have been previously extracted in oral/head and neck cancer were found to be associated with age, ultraviolet and deamination by the APOBEC family [13, 14]. In addition, we identified a signature that is reported for the first time in oral cancer, although it has been previously found in other types of cancers, such as oesophageal and nasopharyngeal cancer. This signature, characterized predominantly by C>T transitions at an NpCpG sequence context and C>A transversions at CpCpC, has been attributed to defective DNA mismatch repair [13, 14].
Further, 120 OSCC tumors were classified into four groups (mutational signature cluster, MSC1-4) and a few singletons based on the intensity of the mutational signatures operative in each tumor using an unsupervised hierarchical clustering method (Fig. 1B). Tumors in the MSC3 group predominantly presented at the early stage (p=0.003), with a minor tumor size (p=0.03) and no spread to regional lymph nodes (p=0.027) (Fig. 1C). The proportion of late-stage tumors in MSC1, characterized by frequent ELAVL1 mutations (p=2.45×10-4), was high (70%) but not significantly different from that for the other groups. The MSC2 group exhibited a low mutation rate (p=2.8×10-5), a late histological stage (p=0.029), and high nodal involvement (p=0.03), while the age of patient at disease onset in this group was younger (p=0.029). In addition, the MSC4 group was associated with recurrent FAT1 and CASP8 mutations (p=0.001 and p=1.2×10-4, respectively). Among the tumors categorized as singletons, three tumors characterized by enrichment of signature 7 were highly mutated and resected from the lip, concordant with previous observations that this signature was associated with exposure to ultraviolet and primarily found in cancers of the lip and skin [13]. These findings suggest that classification of OSCC by the contributions of specific mutational signatures reflects demographic and prognostic correlation.
Significantly and frequently altered genes in OSCC
In previous genomic analyses, genomic aberrations of OSCC were shown to be dominated by tumor suppressor genes that bear a greater number of inactivating mutations than expected by chance [10]. Here, in addition to MutSigCV [21], we used two additional methods, IntOGen [22] and a χ2-based method [23], to identify cancer driver genes with an inactivating mutation bias. Eleven genes were significantly and frequently mutated (q value < 0.1; ≥5% of OSCC) (Fig. 2, Table 1, and Fig. S4-5): TP53 (43%; that is, 43% of tumors with TP53 mutations), FAT1 (35%), EPHA2 (15%), CDKN2A (17.5%), NOTCH1 (29.2%), CASP8 (23.3%), HRAS (11.7%), RASA1 (5%), PIK3CA (17.5), CHUK (5%), and ELAVL1 (6.7%). Among these genes, CHUK and ELAVAL1 mutations have not been previously associated with OSCC. ELAVL1 encodes an RNA-binding protein that binds and stabilizes the transcripts of many cancer-related genes [27]. The protein encoded by CHUK is a serine/threonine protein kinase that is involved in nuclear factor κB (NF-κB) activation and functions as a tumor suppressor through controlling the turnover of cyclin D1 [28, 29]. We observed activation of NF-κB and overexpression of cyclin D1 in tumors harboring CHUK mutations (Fig. S6), indicating that dysregulation of NF-κB signaling is implicated in driving oral tumorigenesis.
Figure 1
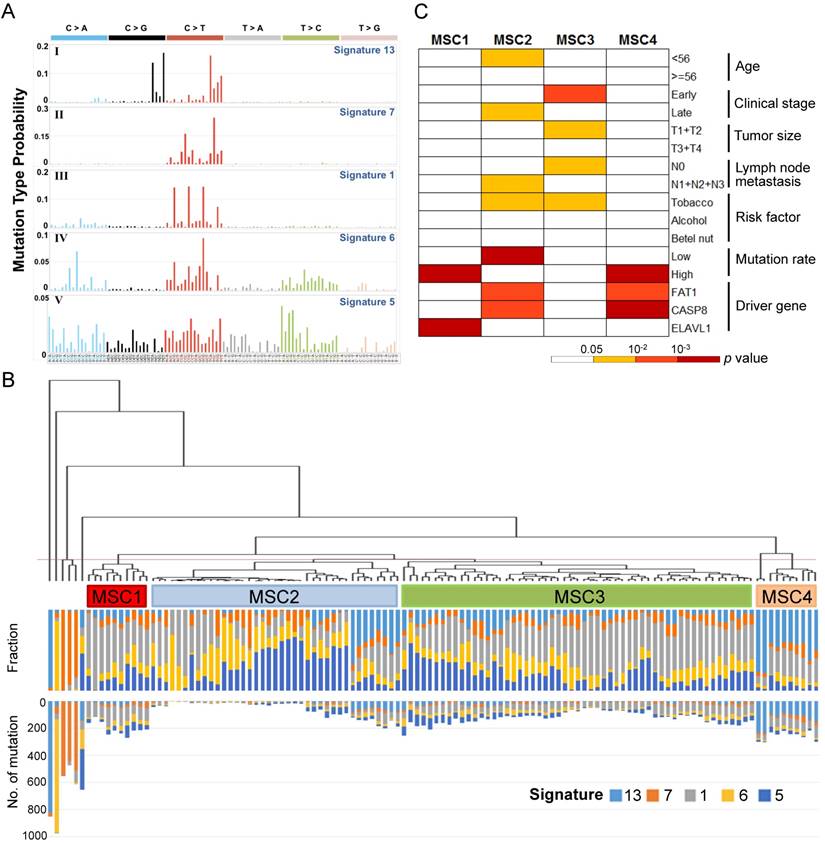
Mutational signatures of 120 oral cancers (A) Five mutational signatures were deciphered in the OSCC cohort and corresponding to the updated consensus signatures [13]. Mutation types are shown in different colors on the horizontal axis. The vertical axis illustrates the percentage of mutations attributed to a specific mutation type. (B) Tumor classification (mutational signature cluster, MSC) based on the contributions of mutational signatures. The fraction and number of mutations attributed to each signature in each tumor is represented by the colored bars below the dendogram. (C) Clinicopathological and etiological characteristics associated with each MSC group.
Figure 2
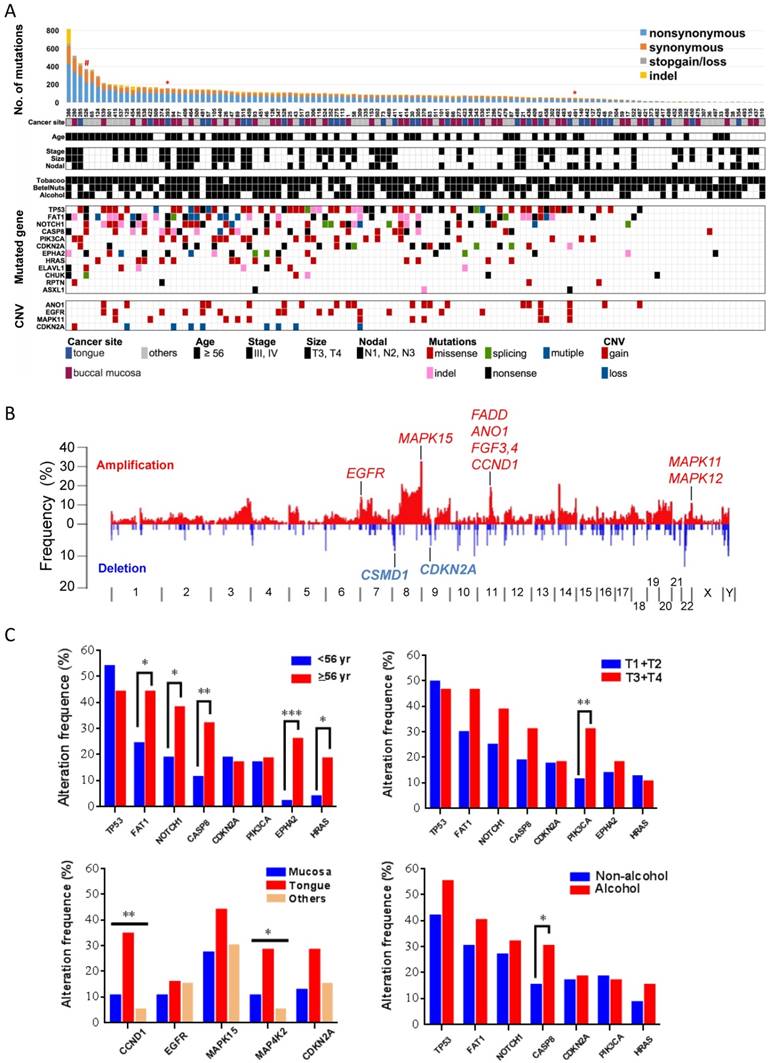
Significantly altered genes in OSCC (A) Mutation rate, demographic and clinical features, environmental exposures and landscape of genomic alterations of 120 OSCC patients. Patients exposed to environmental risks are indicated as a filled square. *, subjected to whole-genome sequencing; #, germline POLD1 mutation. (B) Frequency of SCNAs along the genome. The vertical axis indicates the frequency of SCNAs in the cohort. Selected genes in recurrent amplifications and deletions are labeled. (C) Correlation of gene alteration status with demographic and clinical parameters. *p < 0.05, **p < 0.01, ***p < 0.001, χ2 test for trends in proportion.
Table 1
Genes significantly mutated in OSCC by multiple methods.
| Gene | MutSigCV (q value) | intOGen (q value) | Chi-Square (q value) | Patients number (%) |
|---|---|---|---|---|
| TP53 | 0* | 0* | 2.60666E-21* | 52 (42.5%) |
| FAT1 | 0* | 0* | 0* | 42 (35%) |
| EPHA2 | 0* | 0* | 8.83024E-40* | 18 (15%) |
| CDKN2A | 0* | 0* | 5.60405E-38* | 21 (17.5%) |
| NOTCH1 | 0* | 0* | 2.27504E-21* | 35 (29.17%) |
| CASP8 | 0* | 0* | 7.01792E-12* | 28 (23.33%) |
| HRAS | 0* | 1.79995E-14* | 0.779955677 | 14 (11.67%) |
| CHUK | 0* | 1.01941E-09* | 2.892E-07* | 6 (5%) |
| RASA1 | 0* | 0.000329917* | 2.892E-07* | 6 (5%) |
| ELAVL1 | 0* | 0.000715* | 0.000015424* | 8 (6.67%) |
| PIK3CA | 0* | 0.444915251 | 0.779955677 | 21 (17.5%) |
| RPTN | 0* | ND | 0.779955677 | 4 (3.33%) |
| ASXL1 | 4.73E-05* | 0* | 6.86368E-21* | 3 (2.5%) |
| AHNAK | 0.1394139 | 0.432185662 | 0.779955677 | 4 (3.33%) |
| CHRNB4 | 0.01711482* | ND | 0.019808113* | 4 (3.33%) |
| PEG3 | 0.1213081 | ND | 0.779955677 | 8 (6.67%) |
| NHSL1 | 0.1347749 | 0.002388476* | 9.98024E-05* | 4 (3.33%) |
| F2RL1 | 0.1347749 | 0.05191284* | 9.98024E-05* | 2 (1.67%) |
| VEZF1 | 0.1347749 | 0.432185662 | 0.019808113* | 4 (3.33%) |
| PPM1K | 0.1347749 | 0.467927377 | 0.779955677 | 3 (2.5%) |
| TRIM67 | 0.1394139 | ND | 0.019808113* | 4 (3.33%) |
| PKP3 | 0.2192678 | ND | 0.095549499* | 2 (1.67%) |
| PDHA2 | 0.3252142 | ND | 0.779955677 | 5 (4.17%) |
| H2BFWT | 0.3399323 | ND | 0.265724352 | 2 (1.67%) |
| TMEM165 | 0.3457312 | 0.05191284* | 0.779955677 | 2 (1.67%) |
| PTPN14 | 0.3468882 | 0.002186196* | 4.32573E-07* | 3 (2.5%) |
| PPM1D | 0.3468882 | 0.05191284* | 9.98024E-05* | 2 (1.67%) |
| ATG4C | 0.3707801 | 0.077115199* | 0.007211938* | 3 (2.5%) |
*Statistically significant genes, q<0.1.
Several genes that were frequently mutated showed no significant enrichment in our cohort, including genes either encoding extremely large proteins or spanning an immense genomic region (Table S7). PCLO (16.7%) and MLL3 (8.3%), whose corresponding proteins mediate calcium signaling and epigenetics, respectively, were commonly mutated in many squamous cell cancers [9, 11, 30]. USH2A (8.3%) was identified as a putative tumor suppressor in a genomic analysis of liver cancer [31]. In addition, TTN (28.3%), MUC16 (12.5%), MUC5B (10.8%), MUC4 (9.2%), and CSMD3 (14.2%) were also mutated recurrently in other cancer genome studies [21, 32-35]; however, their roles in driving tumorigenesis remain debated and require further investigations.
We also detected somatic copy-number alterations (SCNAs) by comparing sequence coverage in the 120 tumors and matched non-tumor specimens. The pattern of broad copy-gains and -losses was identified (Fig. 2B), and significantly recurrent SCNAs were detected by GISITC2.0 [25] (Fig. S7). We found recurrently amplified regions on 11q13.3, 7p11.2, 11q22.2, and 22q13.33. Candidate genes in 11q13.3, including CCND1, FGF4, FGF3, and ANO1, are among the most significantly recurrent SCNAs in OSCC. The protein encoded by ANO1 is a voltage-sensitive calcium-activated chloride channel that was recently found to promote cancer progression through functionally interacting with EGFR [36], as we also identified recurrent EGFR amplification in 7p11.2. In addition, consistent with previous reports in oral cancer [10, 11], recurrent copy-loss of candidate tumor suppressors, CSMD1 and CDKN2A was reproducible in our analysis.
Further analyses of gene alteration status with demographic and clinical parameters indicated a significant correlation of age with mutation statuses of many driver genes (Fig. 2C). PIK3CA mutations tended to appear in tumors with large size and CASP8 mutations were more frequently detected in tumors from patients who habitually consumed alcohol. Copy-number alterations in several regions containing CCND1-ANO1 and MAP4K2-VEGFB were substantially enriched in tumors of the tongue.
Since each tumor acquires multiple altered events, we identified two major clusters of associated alterations, separated by HRAS (Fig. 3 and Fig. S8). One cluster is centered on TP53 accompanied with other central regulators of cell cycle, such as CCND1 and CDKN2A. The other mainly comprises a robust interaction among FAT1, CASP8, EPHA2, and NOTCH1. Intriguingly, aberrations of genes characterized to relevant cellular functions/pathways were scattered in the same cluster to a certain degree (Fig. 3A), indicating a joint impact of functional paralogs on oral tumorigenesis.
Figure 3
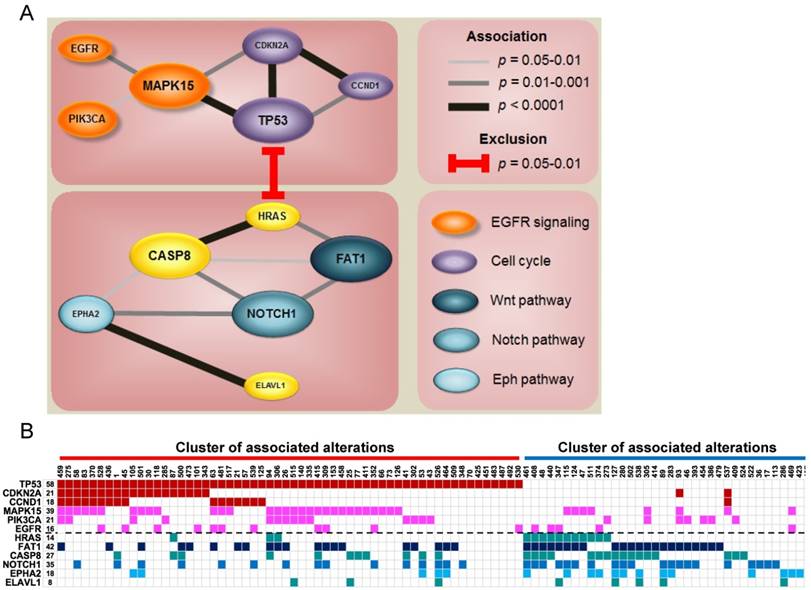
Co-occurrence of gene alterations reveals major groups of associated aberrations. (A) The size of gene indicates the alteration frequency. Significant associations between genes are represented with the darkness of width of the lines. Colors for each gene signify its involvement in a particular pathway. (B) Heat map of altered groups of genes in 120 OSCC.
Actionable events in OSCC
Next, we attempted to define the catalog of targetable genomic alterations in our cohort. We incorporated significantly and frequently mutated genes (q value < 0.1; ≥5% of OSCC) into our list of curated candidates. Also included were genes that can be targeted and were frequently amplified (≥5% of OSCC) (Fig. S9) because it is a real challenge to identify the driver targets of recurrent SCNAs (which often encompass many genes). We empirically annotated these major candidates and found four druggable pathways: mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)-Akt, p53/cell cycle, and Wnt/Notch signal (Fig. 4 and Fig. S10-13). Of these genes, 28 are therapeutic targets of an agent either approved for cancer treatments or evaluated in phase I-III clinical trials. It should be noted that instead of activating mutations, the majority of NOTCH1 mutations in OSCC were predicted to be inactivating (truncated) (Fig. S4), although clinical trials of Notch pathway inhibitors in patients with solid tumors have been reported [37]. Additional Notch-related genes that were frequently altered but not yet targetable are TP63 (9%) and JAG2 (12%). Collectively, 58% of tumors harbored at least one aberrant event potentially targeted by an US Food and Drug Administration (FDA)-approved drug, and 76% carried an actionable alteration by a drug studied in phase I-III clinical trials (Fig. S9). Among these possibly druggable tumors, a great number exhibited two and more targets. As co-occurrence of targetable genes in each sample was observed (Fig. 3 and Fig. S8), if applying a combination therapy specifically directed against the p53-cell cycle pathway (TP53 and CCND1), 44 % were likely targetable, and these tumors are predominantly located in the tongue (Fig. S11 and Fig. S14), indicating differential efficacies of current targeted therapies on anatomical site-specific OSCC.
Figure 4
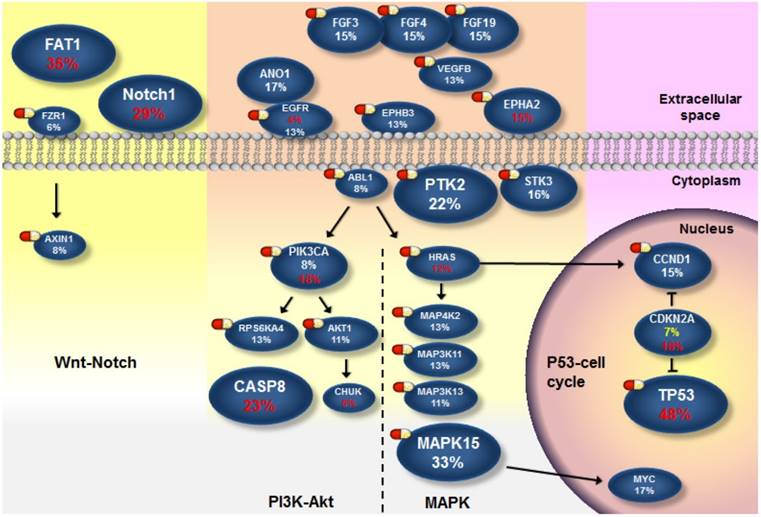
The landscape of major altered genes and potentially druggable pathways in OSCC. Pills show targets of pharmaceuticals (FDA-approved agents or drugs screened in clinical trials). Genes assigned to each pathway are represented with their alteration frequencies (red font, protein-altering mutation; while font, amplification; yellow font, copy loss), and promotive or inhibitory regulations between genes are highlighted.
Discussion
One of the challenges in OSCC studies relates to the myriad of anatomical sites that comprise this disease. In the present study, we showed molecular OSCC subgroups that are associated with clinicopathological parameters and etiological backgrounds according to the intensity of mutational patterns in oral cancer. We also unraveled the spectrum of the major druggable targets/pathways, which will be helpful to identify OSCC patients who could likely benefit from targeted therapies in future clinical trials.
Our analysis identified novel driver genes for oral cancer, one of which, CHUK, encodes the kinase IKKα that blocks NF-κB activation [29]. Aberrant or constitutive NF-κB activation has been detected in many human malignancies, yet oncogenic mutations in NF-κB genes are rare and only occur in its upstream signaling components [38]. Our finding for the first time implicates NF-κB as a therapeutic target for OSCC treatment. In addition, ELAVL1 encodes a member of the Hu family of RNA-binding proteins (HuR) possessing broad cancer-related functions mainly by promoting or repressing translation of various oncogenes and tumor suppressor genes [39]. Of note, while oncogenic functions of HuR have been recognized in many cancer types, a majority of ELAVL1 mutations (5 out of 8) detected here were predicted to truncate the gene product (Fig. S4-5). This suggests that ELAVL1 may act in a tissue-specific manner and function as a putative tumor suppressor rather than an oncogene in OSCC as does NOTCH1. Two significantly but less frequently mutated genes are ASXL1 and RPTN (Table 1). ASXL1, encoding a transcription factor, was frequently mutated in myelodysplastic syndromes and other myeloid malignancies [40]. RPTN, CHUK, and ELAVL1 are all clearly related to epithelial differentiation [28, 41, 42], although a tumorigenic role for RPTN remains undefined.
Notably, we detected strong associations between gene aberrations and revealed altered groups of genes sharing a certain degree of functional correlation (Fig. 3 and Fig. S8). A key group is composed of gene alterations in many vital regulators of cell cycle, such as TP53, CCND1, and CDKN2A. Another group of tumors predominantly accumulate mutations in several major OSCC driver genes including FAT1, CASP8, EPHA2 and NOTCH1. FAT1 is a protocadherin protein that is reported to bind to β-catenin and regulate Wnt signaling [43]. Cross-talking of Wnt/β-catenin, Notch, and EphA2 signaling is central to epithelial differentiation and proliferation [44-46]. Instead of the lethality of gene combinations, our data pinpoint a synergistic contribution of related major drivers to oral tumorigenesis and reflect novel insights into targeted combination therapies to address this fatal disease.
A variety of studies have used high-throughput genomic analyses to characterize the HNSCC and OSCC [8-12, 47]. Despite the presence of a certain degree of consistency, such as recurrent mutations in HRAS and PIK3CA, inactivating mutations in NOTCH1, amplification of EGFR and CCND1, and loss of CDKN2A, differences between these datasets and findings of this study emerge. In comparison with the TCGA head and neck study [12], by far the largest genomic analysis of HNSCC whose cohorts are mostly Caucasian, the alteration frequency of TP53 here (43%) is way lower (84% in TCGA) although TP53 remains the most recurrently mutated gene in our cohort. As such, loss of chromosome 3p, which has been shown to be co-occurring with TP53 mutations in HNSCC [48], is less frequent in our study. In addition, the prevalence of gain of chromosome 3q, another common copy-number alteration encompassing PIK3CA and TP63 in the TCGA study (34%), is not strikingly high in our dataset (8%). Noteworthily, the most common anatomical site of oral cancer in Taiwan is the buccal mucosa (~40%); however, carcinoma of buccal mucosa is relatively rare in the TCGA patients as OSCC mainly presents as tongue cancer in the West (~65%). Furthermore, in addition to tobacco and alcohol use, betel nut chewing is a prevailing risk factor for oral tumorigenesis in Taiwanese males (80%) but very unusual in Western countries. Such variations in the anatomical locations and etiological parameters of this disease associated with the unique cultural practice and life style of different ethnic populations, to some degree, account for the observation that our results are more compatible with data reported from the India Project Team [11] than with that of the Western studies. These regional discrepancies, together with the differences in genomic landscape revealed in the present study reflect a long-standing conjecture that Asian OSCC, especially for those caused by betel nut chewing, is a different disease to the one in the West.
Oral cancer is the fourth leading cause of death due to malignancies in Taiwan [4]. Combination of surgery, radiotherapy or chemotherapy remains the standard treatment of choice for this disease. An anti-EGFR agent in combination with radiotherapy has demonstrated modest benefit for patients with HNSCC, while the activity of EGFR-targeting therapies alone was limited [49]. As a matter of fact, EGFR amplification may not be as prevalent as previously described [50] (13% in our cohort). The abundance of major genomic aberrations in MAPK (66%) and PI3K-Akt (48%) pathways downstream of EGFR compensates each other, likely rendering intrinsic resistance (Fig. 4). On the other hand, TP53, the most frequently altered gene (43%), becomes another appealing therapeutic target with the success for the use of small molecules acting to restore p53 function in clinical trials [51]. In our specimens, we found that TP53 mutation is highly permissive for CCND1 amplification (Fig. 3 and Fig. S8). Recently, a phase I dose-escalation trial of the cyclin D modulator in patients with lymphoma is ongoing after demonstrating promising in vitro and in vivo data in mantle cell lymphoma [52], which potentiates the design of combination therapy directed at p53-cell cycle signaling. In addition, genomic alterations in components of the Wnt pathway were observed in our study (Fig. S13). Intriguingly, inactivation of FAT1 (35%, the second most frequently mutated gene) has been shown to promote Wnt signaling and tumorigenesis [43]. In addition, head and neck cancer cell lines with loss-of-function mutations in the Notch signaling showed a high response rate to a specific inhibitor of the Wnt signaling, suggesting a cross-talk between Notch and Wnt signaling in the development of HNSCC [53]. Our data, together with others' indicate that there should be a shift of emphasis for novel targeted therapies of OSCC.
To define the genomic landscape of OSCC and further explore its clinical utility, additional work is needed to address several limitations of the present study. One issue is that miscellaneous non-coding or structural variants that cannot be detected by exome sequencing contribute to crucial roles in cancer development and therapies [54]. We applied whole-genome sequencing of DNA from two of our samples and identified somatic structural variations in OSCC (Fig. S15). A large-scale analysis of oral cancer using whole-genome sequencing will further extend our knowledge of OSCC biology. In addition, we used DNA from whole blood samples rather than histologically normal oral tissues to eliminate germline variations; however, it is still susceptible to detecting false positives which are irrelevant to cancer development. Another drawback is that the survival data were lacking in this study because a vast number of our patients were enrolled in recent years. This hindered our examinations on clinical relevance as several clinicopathological parameters, such as tumor stage, grade, and nodal involvement were evaluated. Also unavailable was a completed collection of HPV typing data since betel nut chewing was recognized as a prevailing risk of OSCC among males in Taiwan. Infrequent TP53 mutations and a lower mutation rate have been noticed in HPV-positive HNSCC [8, 9], whereas none of these observations was reproducible from Indian populations in a genomic analysis of site-specific OSCC [11]. As the problem of reliable HPV detection remained an issue, a potential utility of massively parallel sequencing to detect not only human but also HPV DNA in tumor specimens has been proposed [9]. These need to be pursued in our future investigations.
Overall, the landscape of genetic alterations in oral cancer mirrors the etiological and medical implications of this tumor type. Major driver candidates/pathways not previously identified in OSCC, such as Wnt and NF-κB, have emerged as potential targeted treatment options from the present study. For patient care, integration of genomic profiles with clinical parameters facilitates translating OSCC genomes into the understanding of oral tumorigenesis and the development of novel therapies for this devastating cancer.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Tissue Bank and Medical Research Center Microscope Room at Chang Gung Memorial Hospital, Keelung for sample preparation and imaging, respectively. This study was supported by research grants from the Ministry of Science and Technology, Taiwan (NSC-102-2314-B-040-008-MY3) and from Chung Shan Medical University Hospital, Taiwan (CSH-2016-E-001-Y2) to SFY, and from Chang Gung Memorial Hospital (CMRPG2F0421) to SCS.
Competing interest
The authors declare that they have no competing interests.
References
1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T. et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71-96
2. Zini A, Czerninski R, Sgan-Cohen HD. Oral cancer over four decades: epidemiology, trends, histology, and survival by anatomical sites. J Oral Pathol Med. 2010;39:299-305
3. Krishna Rao SV, Mejia G, Roberts-Thomson K, Logan R. Epidemiology of oral cancer in Asia in the past decade-an update (2000-2012). Asian Pac J Cancer Prev. 2013;14:5567-77
4. Su CC, Yang HF, Huang SJ, Lian Ie B. Distinctive features of oral cancer in Changhua County: high incidence, buccal mucosa preponderance, and a close relation to betel quid chewing habit. J Formos Med Assoc. 2007;106:225-33
5. Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-8
6. Lee LA, Huang CG, Tsao KC, Liao CT, Kang CJ, Chang KP. et al. Human Papillomavirus Infections are Common and Predict Mortality in a Retrospective Cohort Study of Taiwanese Patients With Oral Cavity Cancer. Medicine (Baltimore). 2015;94:e2069
7. Scully C, Field JK, Tanzawa H. Genetic aberrations in oral or head and neck squamous cell carcinoma (SCCHN): 1. Carcinogen metabolism, DNA repair and cell cycle control. Oral Oncol. 2000;36:256-63
8. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ. et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154-7
9. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A. et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157-60
10. Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM. et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013;3:770-81
11. India Project Team of the International Cancer Genome C. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat Commun. 2013;4:2873
12. Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576-82
13. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-21
14. Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15:585-98
15. Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S. et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618-22
16. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471-4
17. Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246-59
18. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
19. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213-9
20. Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811-7
21. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214-8
22. Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A. et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013;10:1081-2
23. Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE. et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. 2014;20:6582-92
24. Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G. et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28:423-5
25. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41
26. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
27. Wang J, Guo Y, Chu H, Guan Y, Bi J, Wang B. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int J Mol Sci. 2013;14:10015-41
28. Descargues P, Sil AK, Karin M. IKKalpha, a critical regulator of epidermal differentiation and a suppressor of skin cancer. EMBO J. 2008;27:2639-47
29. Kwak YT, Radaideh SM, Ding L, Li R, Frenkel E, Story MD. et al. Cells lacking IKKalpha show nuclear cyclin D1 overexpression and a neoplastic phenotype: role of IKKalpha as a tumor suppressor. Mol Cancer Res. 2011;9:341-9
30. Dotto GP, Rustgi AK. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell. 2016;29:622-37
31. Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z. et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422-33
32. Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609-15
33. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC. et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400-4
34. Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A. et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502-6
35. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G. et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153-8
36. Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S. et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci U S A. 2013;110:E1026-34
37. Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling-are we there yet? Nat Rev Drug Discov. 2014;13:357-78
38. DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246:379-400
39. Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA. 2010;1:214-29
40. Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N. et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145:788-800
41. Huber M, Siegenthaler G, Mirancea N, Marenholz I, Nizetic D, Breitkreutz D. et al. Isolation and characterization of human repetin, a member of the fused gene family of the epidermal differentiation complex. J Invest Dermatol. 2005;124:998-1007
42. Liu L, Ouyang M, Rao JN, Zou T, Xiao L, Chung HK. et al. Competition between RNA-binding proteins CELF1 and HuR modulates MYC translation and intestinal epithelium renewal. Mol Biol Cell. 2015;26:1797-810
43. Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan S. et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet. 2013;45:253-61
44. Crosnier C, Stamataki D, Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet. 2006;7:349-59
45. Nakamura T, Tsuchiya K, Watanabe M. Crosstalk between Wnt and Notch signaling in intestinal epithelial cell fate decision. J Gastroenterol. 2007;42:705-10
46. Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462-75
47. Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker T. et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015;21:632-41
48. Gross AM, Orosco RK, Shen JP, Egloff AM, Carter H, Hofree M. et al. Multi-tiered genomic analysis of head and neck cancer ties TP53 mutation to 3p loss. Nat Genet. 2014;46:939-43
49. Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB. et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567-78
50. Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24:2666-72
51. Parrales A, Iwakuma T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front Oncol. 2015;5:288
52. Prasad A, Shrivastava A, Papadopoulos E, Kuzontkoski PM, Reddy MV, Gillum AM. et al. Combined administration of rituximab and on 013105 induces apoptosis in mantle cell lymphoma cells and reduces tumor burden in a mouse model of mantle cell lymphoma. Clin Cancer Res. 2013;19:85-95
53. Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T. et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110:20224-9
54. Ding L, Wendl MC, Koboldt DC, Mardis ER. Analysis of next-generation genomic data in cancer: accomplishments and challenges. Hum Mol Genet. 2010;19:R188-96
Author contact
Corresponding author: Dr. Shun-Fa Yang, Institute of Medicine, Chung Shan Medical University, 110 Chien-Kuo N. Road, Section 1, Taichung 402, Taiwan; Telephone: +886-4-24739595 ext. 34253; Fax: +886-4-24723229; E-mail: ysfedu.tw.
Received 2016-11-29
Accepted 2017-1-2
Published 2017-2-26
Citation styles
APA
Su, S.C., Lin, C.W., Liu, Y.F., Fan, W.L., Chen, M.K., Yu, C.P., Yang, W.E., Su, C.W., Chuang, C.Y., Li, W.H., Chung, W.H., Yang, S.F. (2017). Exome Sequencing of Oral Squamous Cell Carcinoma Reveals Molecular Subgroups and Novel Therapeutic Opportunities. Theranostics, 7(5), 1088-1099. https://doi.org/10.7150/thno.18551.
ACS
Su, S.C.; Lin, C.W.; Liu, Y.F.; Fan, W.L.; Chen, M.K.; Yu, C.P.; Yang, W.E.; Su, C.W.; Chuang, C.Y.; Li, W.H.; Chung, W.H.; Yang, S.F. Exome Sequencing of Oral Squamous Cell Carcinoma Reveals Molecular Subgroups and Novel Therapeutic Opportunities. Theranostics 2017, 7 (5), 1088-1099. DOI: 10.7150/thno.18551.
NLM
Su SC, Lin CW, Liu YF, Fan WL, Chen MK, Yu CP, Yang WE, Su CW, Chuang CY, Li WH, Chung WH, Yang SF. Exome Sequencing of Oral Squamous Cell Carcinoma Reveals Molecular Subgroups and Novel Therapeutic Opportunities. Theranostics 2017; 7(5):1088-1099. doi:10.7150/thno.18551. https://www.thno.org/v07p1088.htm
CSE
Su SC, Lin CW, Liu YF, Fan WL, Chen MK, Yu CP, Yang WE, Su CW, Chuang CY, Li WH, Chung WH, Yang SF. 2017. Exome Sequencing of Oral Squamous Cell Carcinoma Reveals Molecular Subgroups and Novel Therapeutic Opportunities. Theranostics. 7(5):1088-1099.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.