Abstract
Type 2 diabetes mellitus is a common metabolism disorder characterized by high glucose in the bloodstream, especially in the case of insulin resistance and relative insulin deficiency. Nowadays, it is very common in middle-aged people and involves such dangerous symptoms as increasing risk of stroke, obesity and heart failure. In Vietnam, besides the common treatment of insulin injection, some herbal medication is used but no unified optimum remedy for the disease yet exists and there is no production of antidiabetic drugs in the domestic market yet. In the development of nanomedicine at the present time, drug design is considered as an innovative tool for researchers to study the mechanisms of diseases at the molecular level. The aim of this article is to review some common protein targets involved in type 2 diabetes, offering a new idea for designing new drug candidates to produce antidiabetic drugs against type 2 diabetes for Vietnamese people.
Content from this work may be used under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Type 2 Diabetes mellitus, known as non-insulin-dependent diabetes mellitus or adult-onset diabetes is defined as a metabolism disorder. Metabolism refers to the way our bodies digest food for energy, which in this case means the glucose level in the bloodstream. Diabetes is characterized by high glucose in the bloodstream, especially in the case of insulin resistance and relative insulin deficiency. Type 2 diabetes is a very common disease, which occurs mostly in middle age, but the genes involved have not yet been discovered. It shows significant increased risk of stroke and heart failure, with many additional symptoms such as renal failure, blindness and amputation. Normally patients are treated with diet, drugs and most commonly, insulin injection. Many new generations of drugs have been produced in silico, gaining their specificity and efficacy acting as active biomolecules interacting with the target sites in human body molecular complex structures. Current type 2 diabetes drugs are not only expensive but also reveal several significant side-effects. In Vietnam, however, diabetes treatment medication has mostly been derived from herbal sources. Most herbal sources used are from the traditional medication of ancient times, such as bittermelon (Momordica) [1], Giao Co Lam (Gynostemma pentaphyllum) [2], Tho Phuc Linh (Smilax Glabra) [3, 4], Okra (Abelmoschus escunlentus) [5], Luc Vi Dia Hoan (named phanoside) [6], etc. These medicines can be used separately or conjointly in a complete remedy. Recently there has been some usage of methi seed (fenugreek) imported from India. But still there are no unifying remedies for type 2 diabetes specifically from herbal-derived medication, and there is still no report at protein or molecular levels on the action of these drugs on protein target in the mechanism of deactivation of the disease. According to a report at the Cardiology Conference in Ho Chi Minh City in 2004, 75% of diabetes patients died because of stroke, 55% of them due to coronary thrombosis, and more than 5% of adults have type 2 diabetes. However, antidiabetic drug treatment can still not be produced widely in the domestic market because of the lack of scientific knowledge guidance.
In general, greater efforts are needed in the design, characterization and application of materials and devices at nanoscale for treating the disease. In this article, we review all potential protein target sites type 2 of diabetes disease and elucidate its mechanism. The aim of our work is to provide for opportunities to strategically develop effective treatment [7].
2. Mechanisms involved in the disease and common protein target
The search for the optimal treatment of type 2 diabetes might have complications due to the complex pathogenesis of the disease. There are many searches for therapy to improve the action of insulin on its target tissues and finding those compounds which have the ability to improve the secretion of insulin by beta-cells. In the past decade, several new oral agents have been discovered to control blood sugar for type 2 diabetes patients. These pharmacological agents work by different mechanisms and with adverse effects. There are the following common groups.
2.1. Insulin secretagogues
These can be classified as drugs designed to delay the absorption of carbohydrates from the gastrointestinal tract and insulin sensitivity. Commonly there are two kinds of secretagogue drugs: sulfonylureas and non-sulfonylureas. Sulfonylurea drugs are those agents stimulating pancreatic beta-cell insulin secretion, such as glibenclamide (glyburide), glipizide, chlorpropamide, tolbutamide and glimepiride. Non-sulfonylurea insulin secretagogues are newer agents that also stimulate insulin secretion but are more short-acting. Figure 1 illustrates schematic representation of the triggering and amplifying pathways involved in the control of insulin secretion by glucose. It is supposed that there are six potential sites of action of insulin-secreting drugs.

Figure 1 Potential six sites of action for insulin-secreting drug.
Jean-Claude Henquin [8], from American Diabetes Association, proposed six potential sites of action for insulin-secreting drugs in 2004, which are summarized in table 1.
Table 1. Pathways of potential action of insulin-secreting drugs.
| Site 1 |
Stimulation of
 -cell metabolism
-cell metabolism
|
| Activation of glucokinase | |
| Inhibition of glucose-6-phosphatase | |
| Alternatives fuels | |
| Inhibition of mitochondrial Na +/Ca 2+ exchanger | |
| Site 2 |
Increase of
 -cell
[Ca2+
]i
by blockade of
KATP
channels
-cell
[Ca2+
]i
by blockade of
KATP
channels
|
| Interaction with SUR1 | |
| Interaction with K + IR 6.2 | |
| Site 3 | Increase of [Ca2+ ]i by action at sites |
| other than KATP channels | |
| Blockade of other K + channels | |
| Activation of Ca 2+ channels | |
| Activation of ionic channel | |
| Inhibition of [Ca 2+] i lowering processes | |
| Site 4 |
Stimulation of amplifying pathways in
 -cells
-cells
|
| Activation of the nutrient-mediated amplification | |
| Inhibition of AMP kinase | |
| Inhibition of 11β-hydroxysteroid dehydrogenase type 1 | |
| Sensitization to Ca 2+ | |
| Inhibition of cAMP degradation | |
| Activation of the PKC pathway | |
| Site 5 |
Action on
 -cell membrane receptors
-cell membrane receptors
|
| Antagonists of inhibitory receptors | |
| Agonists of stimulatory receptors | |
| Site 6 |
Action on
 -cell nuclear receptors
-cell nuclear receptors
|
The first insulin secretagogues to consider are sulfonylureas. They have been used as the original oral medication for type 2 diabetes patients in the United States since 1955. Sulfonylureas signal insulin release from the pancreatic beta cells by the mechanism on Ca 2+ channels, allowing Ca 2+ to enter the voltage-dependent Ca 2+ channels, increasing the free cytosolic calcium level (figure 2). Hellman et al [9, 10] showed that sulfonylureas together with tolbutanmide, owing to the insulin secretory process by a lipophillic interaction with the phospholipid domain in the membrane, play a role in activating the voltage-dependent Ca 2+ channel. Studies have demonstrated that sulfonylureas bound with a high affinity to specific receptors on the beta-cell membrane reflect the ability of these compounds to stimulate insulin secretion from the beta-cell. They are bound to a receptor located on the plasma membrane adjacent to potassium channels, called sulfonylurea receptor (SUR) whose possible endogenous transmitter is not known. SUR activation inhibits ATP-sensitive K+ channels leading to a reduced efflux of potassium. The rise of intracellular potassium concentration creates a sufficient cellular depolarization to elicit the opening of voltage-dependent calcium channels. This ultimately increases intracellular Ca 2+ which elicits insulin secretion. Sulfonylureas can, moreover, inhibit glucagon secretion and sensitize target tissues to the action of insulin. Sulfonylureas of the second generation—glipizide, glibenclamide, gliclazide, glibornuride and glimepiride, are active with doses much lower than those of the active sulfomylureas of the first generation: tolbutamide, chlorpropamide and carbutamide. However, sulfonylureas and insulin have reduced microvascular risk by 37%, according to the UK Prospective Diabetes Study (UKPDS), but other risks of myocardial infarction, stroke and congestive heart failure also decreased [11].
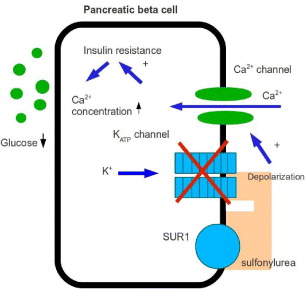
Figure 2 Action of sulfonylurea on β-cell.
2.2. Alpha-glucosidase inhibitors
These drugs act to slow the absorption of carbohydrates, reducing postprandial elevations in plasma glucose level. They cause a rather mild reduction in hemoglobin A1c (HbA1c) but do not show any significant plasma glucose level reduction during the fast. Drugs such as acarbose and miglitol are the most common of this group. The mechanisms of these drugs are based on the action of membrane-bound enzymes, the intestinal alpha-glucosidases in the intestinal brush borde. These enzymes hydrolyze starch residues, oligosaccharides and disaccharides, thus releasing glucose in the intestine brush borde. This hydrolysis is necessary for the absorption of digestive carbohydrates, because only monosaccharides like glucose and fructose can be absorbed. Inhibition of alpha-glucosidases by products such as acarbose, miglitol or emiglitate, reduces and delays the digestive absorption of glucose and decreases post-prandial hyperglycemia [12].
2.3. Dipeptidyl pepdidase 4 (DPP-4) inhibitors
Glucagon like peptide-1 (GLP-1) is one of the important hormones, in response to metabolism. GLP-1, together with glucose-dependent insulinotropic peptide (GIP), released after meal ingestion, delay gastric emptying, increase insulin secretion and reduce glucagon secretion [13]. However, these incretins are degraded easily by dipeptidyl peptidase-4 (DPP-4), and therefore, GLP-1 is no longer the most interesting therapeutic target, though it might be considered as an antidiabetic hormone [14]. DPP-4 is widely expressed on the surface of many cell types of many organs. DPP-4 may circulate in a soluble form [15, 16]. The mechanism of DPP-4 in inactivation of these incretins by cleavage of the two NH2-terminal amino acids of bioactive peptides, forming alanine or proline to the second amino-acids of GLP-1, make it cleaved to a truncated form [16, 17]. Thus, GLP-1 always has a short half-life, less than 2 min, causing many constraints in treatment using the native form of GLP-1. Therefore, the therapeutic strategy of treatment is using DPP-4 inhibitors to slow down the degradation process of incretin; DPP-4 inhibitors increase the effects of GLP-1 and GIP two to three times in both animal models and in patients [16]. The mechanism of DPP-4 inhibition can be described as that DPP-4 inhibitors prevent GLP-1 activation, and thus, it increases GLP-1 levels. Much evidence from animal model studies showed the importance of DPP-4 inhibition on GLP-1 levels and furthermore, on insulin secretion which also improves β-cell.
2.4. Peroxisome proliferator-activated receptor (PPAR-γ)
Recently the development of the safer antidiabetic agent target PPAR-γ was announced by Choi et al [18]. It is the type 2 nuclear receptor, mainly expressed in adipose [19]. PPAR-γ has three messenger ribonucleic acid (mRNA) isoforms. At different levels and positions PPAR-γ1 is predominantly expressed widespread, albeit at low level, while PPAR-γ2 and PPAR-γ3 are expressed more highly in adipose tissue. It is interesting to notice that only adipose tissue expresses the highest level of PPAR-γ mRNA, namely PPAR-γ in skeletal muscle, and it is found to be increased with insulin resistance [20, 21]. PPAR-γ also has its regulation by insulin, tumor necrosis and glucocorticoids [22]. There is also a paradox for PPAR-γ in how its agonist improves insulin sensitivity in muscle in glucose uptake up to maximum, because adipose tissues play important roles in the human body and are responsible for glucose homeostasis. As a typical character of nuclear receptors, PPAR-γ shows up a structure of distinct functional domains and the receptor exerts many of its effects by regulating target gene transcription in a ligand-dependent manner. When PPAR-γ is bound with a ligand or some kind of synthetic compound, such as thiazolidinedione, the whole structure becomes active and complex with another transcription factor retinoid X-receptor (RXR). The heterodimer complex PPAR-γ-RXR is then bound to a specific DNA motif, which is known as peroxisome proliferate response element (PPREs) in the promoters of target gene [23]. This then leads to the activation of regulation. Thiazolidinedione belongs to a class of drugs (glitazones) acting primarily as effective insulin sensitizers, which have specific high-affinity ligands for PPAR-γ. Another remarkable feature of thiazolidione insulin sensitizers is their synergism with other glucose-lowering drugs like metformin and sulfonylurea. Glitazones in combination with metformin, insulin and sulfonylureas, while added to current treatment in patients with glycemic control, are proposed to act effectively.
3. Proteins involved in drugs against type 2 diabetes
The 3D structures of most proteins were obtained by using any of three methods: x-ray diffraction (XRD), nuclear magnetic resonance (NMR) and theoretical modeling. XRD and NMR are two of the most common methods. Since they are experimental methods, the empirical results from them can more accurately describe the 3D structure than the others. By the treatment of NMR data one can derive the molecular parameters of proteins and calculate their 3D structures. From XRD experimental results one can obtain the electron density map and recognize the crystallization or solution phase of the electron clouds. The solvent effect on protein can be examined by observing the crystallization of the same protein into many crystalline forms in different solvents. Therefore, one can get the best whole 3D structure on the basis of the analysis of above-mentioned data. However, the hydrogen atom in this method is hard to examine because it has only one electron. In a newer model of x-ray crystal diffraction, hydrogen atoms can be added to the structure by modeling [24]. The temperature and resolution are the most important factors that affect the final model of the structure [25].
3.1. 11β-Hydroxysteroid dehydrogenase (HSD)
11β-HSD belongs to short-chain dehydrogenase/reductase (SDR) protein family, which catalyzes the conversion of cortisone to cortisol available in brain, liver and adipose tissues in humans. Its isoform 11β-HSD2 is expressed predominantly in liver. Cortisol is an active glucocortisoid, which plays a key role in diabetes, so 11β-HSD is an important therapeutic target for type 2 diabetes.
Many studies have shown that the high circulating levels of the active glucorticoid cortisol lead to insulin-resistant diabetes, central obesity, dyslipidemia and hypertension [26]. 11β-HSD1 inhibition is a tempting target for the treatment of glucortinoid-associated diseases, especially of type 2 diabetes [27, 28]. 11β-HSD1 activity is highly dependent upon the cofactor nicotinamide ademine dinucleotid phosphate/micotinamide ademine dinucleotid phosphate-oxidase (NADP/NADPH), while type 2 is nicotinamide ademine dinucleotid (NAD+)-dependent dehydroductase (figure 3). 11β-HSD1 activity is determined by a near protein, hexose-6-phosphate hydrogenase, which generates NADPH in the endoplasmic reticulum (ER)-lumen [29]. 11β-HSD1 has both dimer and tetramer organizations. Hydrophobic N-terminal determines their orientation in the ER membrane while C-terminal of one subunit caps the active site of the other partner in dimer. Its activity is also affected by glycosylation and membrane attachment [30]. 11β-HSD1 is crystallized to serve various studies on 11β-HSD inhibitors. However, the crystal structure, which is a homodimer in an asymmetric unit, is devoid of hydrophobic N-terminal portion. The 3D structure of 11β-HSD1 is given in table 2.
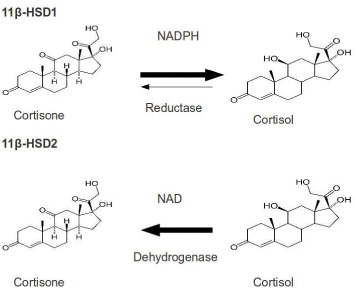
Figure 3 Interconversion of cortisone and cortisol mediated by 11β-HSD1 and 11β-HSD2.
Table 2. 3D structure of 11β-HSD1 according to Protein Data Bank (PDB) code and species using x-ray diffraction method.
| Murine | |||

|

| ||
| PDB ID: 1Y5M; | PDB ID: 1Y5R; | ||
| Resolution: 2.30 Å; | Resolution: 3.00 Å; | ||
| Reference: Zhang [31] | Reference: Zhang [31] | ||
| Guinea pig | |||

| |||
| PDB ID: 1XSE; | |||
| Resolution: 2.50 Å; | |||
| Reference: Ogg [30] | |||
| Human | |||

|

|

| |
| PDB ID: 1XU9; | PDB ID: 1XU7; | PDB ID: 2BEL; | |
| Resolution: 1.55 Å; | Resolution: 1.80 Å | Resolution: 2.11 Å; | |
| Reference: Hosfield [32] | Reference: Hosfield [32] | Reference: Wu et al [33] |
Several attempts have been made to find out possible effects of membrane attachment on 11β-HSD1 activity due to the fact that 11β-HSD1 transcript, lacking hydrophobic N-terminal, can be active only when it is attached to the ER membrane. Its activity becomes very unstable and is lost within hours after solubilization and release from the ER membrane [34]. In vitro 11β-HSD1 and its isomer 11β-HSD2 anchor the ER in opposite directions. Particularly, the N-terminal of this enzyme faces cytosol space and the C-terminal harboring catalytic residues group is directed toward ER-lumen while in the case of 11β-HSD2, the N-terminus is lumenal and the C-terminal is cytoplasmic [29]. The oxidative environment inside the ER-lumen requires cofactor to allow the catalysis of the reductase reaction by 11β-HSD1. The consequence of 11β-HSD1 inhibition also needs to be taken into account since 11β-HSD plays other important roles in regulating oxysterol metabolism [35].
3.2. 17β-Hydroxysteroid dehydrogenase type 1 (17β-HSD1)
17β-HSD1 plays a pivotal role in the local synthesis of the most potent estrogen estradiol. Its expression is a prognostic marker for the outcome of patients with breast cancer. Its inhibition is currently under consideration for breast cancer prevention and treatment.
17β-HSD1 is over-expressed in breast tumor cells, because estrogen target cells 17β-HSD1 catalyze the NADPH-dependent reduction of estrone (E1) to the potent 17β-estradiol (E2) [36]. For women in the post-menopausal phase, the proliferation of this hormone is led by increased levels of E2, so it is commonly considered as a novel therapeutic target (figure 4).
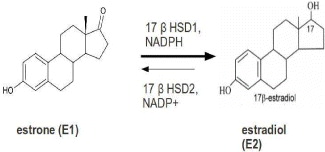
Figure 4 Reaction of the reduction from E1 to E2.
This enzyme is thus an important target for inhibitor design. The structure of recombinant human 17β-HSD1 in complex with estradiol at room temperature has been determined at 1.7 Å resolution, and a ternary 17β-HSD1–estradiol–NADP+ complex at −150 °C has been solved and refined at 2.20 Å resolution [37]. The structures show that estradiol interacts with the enzyme through three hydrogen bonds, and hydrophobic interactions between the core of the steroid and nine other residues (figure 5). Recently, the 3D structure of human 17β-HSD1 has been determined independently at 2.20 Å resolution [38]. From proposed models only in the structure of the ternary complex there are descriptions of substrate interaction at the active site and a putative mechanism [38]. The main characteristic of the packing is the formation of a crystallographic dimer, thought to be equivalent to the active dimer in solution. Twenty crystal structures of 17β-HSD1 had been available in PDB by May 2010.
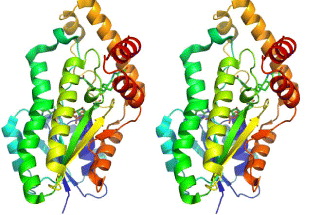
Figure 5 The overall structure of 17β-HSD1–estradiol–NADP+ complex. Stereoscopic view of 17β-HSD1, complexes with estradiol and NADP+.
3.3. Glutamine fructose-6-phosphate amidotransferase (GFAT or GFPT)
GFAT plays a key role in hexosamine biosynthetic pathway, which is involved in glucose-induced insulin resistance and the induction of the synthesis of growth factor [39]. The hexosamine pathway is the sensor of the glycoglysis pathway, in the reaction of fructose-6-phosphate (F6P) with the NH 2 donor amino acid, a glutamine which is converted to glucosamine-6-phosphate (GlcN6P) by the glutamine fructose-6-phosphate amidotransferase (GFA) enzyme. Subsequent reactions result in the formation of uridine diphosphate N-acteylglucosamine (UDP-GlcNAc) which is utilized as a substrate for N- and O-linked glycosylation. It is intracellular protein O-glycosylation mediated by O-linked GlcNAc transferase (OGT) which has been postulated to contribute to glucose toxicity by altering gene expression.
The synthesis of glucosamine begins with the transport of glucose into the cell and its conversion to glucose-6-phosphate by hexokinase and then isomerization to fructose-6-phosphate by the enzyme phosphofructose kinase I of the glycolysis pathway. Then fructose-6-phosphate can go through the normal metabolic pathway as previously described in glucose metabolism. Due to the hexosamine pathway, 2–5% of fructose-6-phosphate formed in glycolysis is converted to glucosamine. Fructose-6-phosphate is isomerized and aminated by glutamine and GFAT (L-glutamine, D-fructose-6-phosphate amidotransferase) to yield glucosamine-6-phosphate [40]. First, acetyl-coenzyme (CoA) from either glucose metabolism or fatty acid oxidation transfers its acetyl group to glucosamine-6-phosphate to produce N-acetylglucosamine-6-phosphate [41, 42]. Then, a uridine nucleotide (UDP) is added to the glucosamine yielding uridine 5'-diphospho-N-acetylglucosamine (UDP-GlcNAc) [40]. Overall the production of UDP-GlcNAc requires glucose, glutamine, acetyl-CoA, uridine, and ATP. Studying the interaction of GFAT with UDP-GlcNAc and its 3D structure will provide more chances to develop a new strategy for treatment of type 2 diabetes. The majority of glucose will enter the glycolysis pathway, with a small percentage entering the hexosamine pathway. Hexosamine biosynthesis is known to contribute to insulin resistance and the induction of the synthesis of growth factor. GFPT or GFAT regulate the hexosamine pathway products. Therefore, this is involved in a therapeutic target against type 2 diabetes [43]. The human GFAT has three isoforms, GFAT1, GFAT2 and GFAT1L [44, 45]. GFAT1 gains most interest because it has high expression in liver and fatty tissues, and therefore is a target against diabetes and obesity. Human GFAT1 has two forms, monomer (figure 6) and dimer (figure 7). UDP-GlcNAc, as mentioned above, is an important ligand to the hexosamine pathway, and the smallest unit of GFATs with full competent characteristics is a dimer, so it is essential to build a 3D structure of a dimer. The template to develop 3D structure of GFAT1 is generated from the crystal structure of E. coli GFAT (GLmS). Its PDB code is 1jax. The human GFAT1 monomer contains two domains: N-domain (residues 1–310) and C-domain (residues 311–680). The complete structure of human GFAT1 monomer has the binding site of glucose-6-phosphate (G6Q) in the C-domain and the ligand binding site of glutamine in the N-domain. The binding site of human GFAT1 with G6Q was shown to have the atomic coordinate which is similar to that in GLmS with 1jax PDB code. The binding site of GFAT1 with glutamine was derived by superimposing the crystal structure (PDB code: 1gdo) on to the structure of GFAT1. The generating method for building the dimer structure for human GFAT1 is similar to that used to build the 3D monomer structure. It can also be done by performing the modeling procedure based on the same sequence alignment but using both B-chain and C-chain of the crystal structure of GLmS as a template. The two chains of the dimer were subjected to being an overall energy minimization to all atoms which finalize the entire structure of the dimer. These findings can gain mutagenesis studies and synthesis compounds as inhibitors for therapeutic treatments of type 2 diabetes.
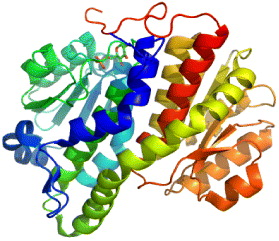
Figure 6 Crystal structure of GFAT1 (monomer).
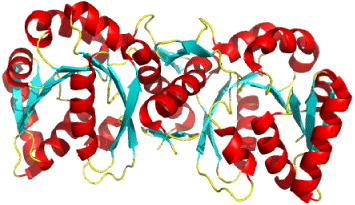
Figure 7 Crystal structure of GFAT1 (dimer).
3.4. Protein tyrosine phosphatase 1B (PTP1B)
In cell system, tyrosine phosphorylation is the process of addition of a phosphate (PO 4) group into a protein or a molecule. Phosphorylation is a very common process because phosphate can regulate many enzymes and receptors, causing or inhibiting the mechanisms of many diseases like type 2 diabetes. Tyrosine phosphorylation is a process rather rare than common; still, its function is in research for cell elongation, proliferation and differentiation. Tyrosine phosphorylation of type 1 insulin-like growth factor (IGF-I) receptor is the autophosphorylation of tyrosine residues in the insulin receptor activation loop. The receptor bound to IGF-I has tyrosine kinase activity and can be bound with a high affinity to activate many cellular proliferations. Tyrosine phosphorylation process is controlled by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). PTPs are a group of enzymes that remove phosphate groups from phosphorylated tyrosine residues on proteins. PTP's family is a largely diverse protein family of enzymes. PTP is recognized by its unique 11-residue sequence motif (I/V) HCXAGXXR(S/T) G with cysteine and arginine essential for catalysis. Recent studies report that PTP1B (figure 8), which is the first PTP to be isolated in homogenous form, plays a role as an important regulator of the insulin-signaling pathway [46].
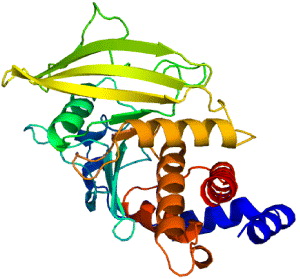
Figure 8 Protein tyrosine phosphatase 1B [PDB code: 3A5J].
The process of insulin signal transduction involves tyrosine phosphorylation in the insulin-receptor activation loop. The insulin receptors are therefore added more, together with the activation of phosphatidylinositol-3-kinase (PI3K) and downstream protein kinase B, activation and subsequent translocation of the glucose transporter type 4 (GLUT4) [47]. This process is regulated by PTP1B by dephosphorylating phosphor-tyrosine residues of the tissue insulin receptor kinase. Being published first in 1994, the crystal structure of catalytic domain of PTP1B is a 37 kDa (residues 1 to 321), which has been determined at 2.8 Å resolution. The catalytic site of the enzyme is located at the base of a shallow cleft. The phosphate recognition site is created from a loop located at the N-terminus of an alpha-helix. This site contains 11-residue sequence motif, which is typical for PTPs, together with catalytically active cysteine and arginine. In this structure, sodium tungstate was used as a heavy metal derivative for its tight binding to PTP1B.
The structure of PTP1B is the framework model for other members of the PTP family and a milestone for understanding of tyrosine dephosphorylation. PTP1B is known to be the first PTP enzyme which was purified from human placental tissue in 1998. In 435 amino-acid composed protein the catalytic domain comprises 30–278 amino-acids. The 35C-terminal residues are rich in proline, playing a role in targeting the enzyme to the cytoplasmic face of the endoplasmic reticulum. The catalytic loops comprise the main structure. They contain the catalytic residue Cys 215, the WPD loop (tryptophan, proline and aspartic acid) and the secondary aryl-phosphate-binding site. The active site of PTP1B is the same as those of all PTPs structures. The base of the catalytic site is defined by 214–221 amino-acids of PTP signature motif [48] which is a loop of 8 amino acid residues coordinated to the phosphate group attached at the 4th position of the phenyl group in tyrosine, the aryl-phosphate moiety of the substrate. Four other loops are rigid. Residues form the sides of the catalytic cleft and contribute to catalysis and substrate recognition process. The depth of the catalytic cleft is about 8–9 Å [46].
Drug design relies on the conformation of 3D structure through XRD/NMR spectroscopy of the protein. PTP1B is a new target for drug design against type 2 diabetes mellitus for its management of insulin resistance. The crystal structure of PTP1B with the inhibitors proclaims that there is another binding site (Arg 24 and Arg 254) besides the phosphotyrosine binding site (residues Cys 215–Arg 221). Inhibitors bound into the two binding sites are found to have high potential in activities at the nanomolar range [46]. Recently, a third binding site was discovered (residues Tyr 46–Asp 48) due to its potential activities and selectivity of inhibitors. The potential of new drug design was to demonstrate that the antisense oligonucleotides to PTP1B reduced the levels of the phosphatase in liver and fat, but not in muscle, which was known to enhance the insulin sensitivity.
3.5. Mono-ADP-ribosyltransferase-sirtuin-6 (SIRT6)
The crystal structure of SIRT6 has 3K35 PDB code (figure 9), which was deposited on 1 October 2009. It has many alternative names such as SIR2L6, NAD-dependent deacetylase sirtuin-6, sirtuin type 6, Sir2-like protein 6, Sir2-related protein type 6. The protein size is 39119.8 Da and the number of residues is 355 amino acids. The function of NAD+-dependent protein deacetylase in biological processes is gene silencing, cell cycle progressing, chromosome segregation, cell apoptosis and survival [49]. There are seven proteins of the sirtuin family (SIRT1 to SIRT7) that share 250 amino acid core domains. The authors solved the crystal structure of human SIRT6 in complex with ADP-ribose at 2.0 Å resolution. Being a mammalian enzyme, SIRT6 is a nuclear protein which is linked to heterochromatic regions. It has been reported to have both ADP-ribosyl transferase and deacetylase activity [50]. SIRT6 belongs to the sirtuin protein family, together with SIRT1–SIRT7, those are homologs to the yeast Sir2 protein. Members of the sirtuin family are characterized by a sirtuin core domain and grouped into four classes. Human sirtuin's function was then unknown, but yeast Sir2 protein was known to affect genomic stability of yeast aging. This protein is a complex that coats telemeres and rDNA repeats, therefore it can promote genomic stability. Blander and Guarente discovered that Sir2 is also a NAD+-dependent deacetylase and life span extension is calorically restricted by yeast [51]. Then several studies at first focused on SIRT1, which is the most correlated homolog of yeast Sir2 [49].
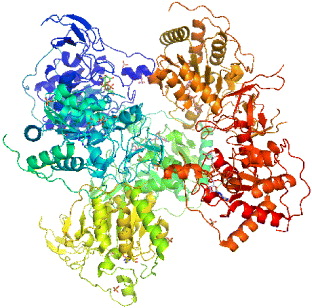
Figure 9 Crystal structure of SIRT6 (PDB code: 3K35).
Mostoslavysky et al [52] studied the ability of SIRT6 to prevent DNA damage and maintain genomic stability. SIRT6 can degrade mouse embryo fibroblasts (MEFs) and undifferentiated stem (ES) cells, showing sensitivity to alkylating and oxidizing agents [52]. SIRT6 was found to have the ability to deacetylate histones and DNA pol β [52]. Therefore, we can conclude that the mechanism of enzymatic activity of SIRT6 is a very interesting issue, as it can serve as a target for drug design against aging disease. Another study involved how cellular enzyme affects blood glucose levels in mice [53] describing an experiment showing that blood sugar can be induced dramatically while enzyme SIRT6 is absent in mice. This led to the new way of learning how this protein can control the way that a cell uptakes glucose, which means we will find a new way to treat type 2 diabetes and even cancer.
4. Discussion about common antidiabetic drugs development
Antidiabetic drugs are medicines which help to control blood sugar levels of people with diabetes mellitus. Patients with type 2 diabetes have to uptake insulin every day to maintain the glucose level in blood because the pancreas produces little or no insulin. For type 2 diabetes treatment, besides insulin injection, four types of medication (oral hypoglycemics) can be used to control the level of glucose. The four types of oral hypoglycemics are sulfonylureas, such as glipizide (glucotrol), glyburide (diabeta, glynase, micronase), chlorpropamide (diabinese) and tolbutamide (orinase); biguanides, such as metformin (glucophage); alpha-glucosidase inhibitors, such as acarbose (precose) and miglitol (glyset); and thiazolidinediones, such as troglitazone (rezulin). But at present, there is no available drug other than insulin considered as interacting directly with insulin receptors. There is much ongoing research to find other non-peptide drugs which are able to activate the insulin receptors. Patients with type 2 diabetes will have to measure glucose in the blood stream by checking the glycated hemoglobin A1c level, in order to achieve an appropriate medication. In a healthy person, normal A1c would be less than 4%, but for people with diabetes, as suggested by the American Diabetes Association (ADA), A1c is 7% or higher [54]. Antidiabetic drugs may refer to several types with specific function; drugs that increase insulin; drugs that increase insulin sensitivity; drugs that decrease glucose influx, drugs decreasing the liver's glucose output and those sequester bile acids.
5. Conclusion
Type 2 diabetes is the metabolism disorder symptom where the body is unable to produce insulin properly to control glucose level in the bloodstream. Prospective clinical studies have proven the benefits of tighter glucose control in reducing the frequency and severity of complications of the disease, leading to the advocacy of earlier and more aggressive use of insulin therapy. Given the reluctance of patients with type 2 diabetes to inject themselves with insulin, orally active insulin mimetics would be a major therapeutic advance. Several proteins have been reported above which are involved in potential drug design for type 2 diabetes, and for further understanding of the mechanism of action of the biology of glycogen metabolism and insulin secretion in order to synthesize several natural compounds to serve as drug candidates. Vietnam, with rich herbal resources, has a great opportunity to develop type 2 diabetes drugs. Our suggestion is to isolate bioactive compounds from known herbals and test their interactions with the selected target proteins presented above for better understanding of those compounds and protein interactions which lay a solid foundation for the development type 2 diabetes drugs.
Note. Figures of all crystal structures are rendered using PyMol, software developed by Warren L DeLano.
Acknowledgment
The work was funded by the Ho Chi Minh Institute for Computational Science and Technology (ICST) through grant number 202/TB-SKHCN on 2 November 2010 for SDR project.